All published articles of this journal are available on ScienceDirect.

Certain Diet and Lifestyle May Contribute to Islet β-cells Protection in Type-2 Diabetes via the Modulation of Cellular PI3K/AKT Pathway
Authors Info & Affiliations
Abstract
PI3K/AKT pathway has been shown to play a pivotal role on islet β-cell protection, enhancing β-cell survival by stimulating cell proliferation and inhibiting cell apoptosis. Accordingly, this pathway appears to be crucial in type-2 diabetes. Understanding the regulations of this pathway may provide a better efficacy of new therapeutic approaches. In this review, we summarize advances on the involvement of the PI3K/AKT pathway in hypothetical intra-cellular signaling of islet β-cells. As recent findings may show the nutritional regulation of the survival pathway in the islet β-cells through activation of the PI3K/AKT pathway, we also review studies on the features of several diets, correlated lifestyle, and its signaling pathway involved in type-2 diabetes. The molecular mechanisms contributing to the disease are the subject of considerable investigation, as a better understanding of the pathogenesis will lead to novel therapies against a condition of the disease.
1. INTRODUCTION
Diabetes mellitus is the most common endocrine disease, which is mainly characterized by pancreatic islet β-cells failure. It is well-known that several stressors like chemical toxins, cigarette smoke, hypertension, and hypercholesterole-mia are all risk factors to the disease [1, 2]. In addition, inflammation is major targets of the diabetes research, which is believed to be the principal reason behind the disease [3]. Diabetes was historically considered as a metabolic disease of just ageing. However, the role of chronic systemic inflammation is important in the development of insulin resistance and following progression to the disease. Now, pathophysiology of type-2 diabetes has identified to be the presence of islet-specific T-cells in the patients [4]. Furthermore, the T-cell-mediated islet autoimmune status has also been correlated with the progressive loss of β-cell function [4]. In general, the quantity of islet β-cells is determined by the combined rates of proliferation of existing cells by neogenesis and by cell death [5]. Reactive oxygen species (ROS), endotoxins, and inflammatory cytokines, result in the decrease of islet β-cells henceforward to the diabetic disease development [6]. Chronic hyperlipidemia also causes β-cells apoptosis and dysfunction, thereby contributing to the pathogenesis of diabetes [7].
Because phosphatidylinositol-3 kinase (PI3K) and serine–threonine protein kinase AKT (also known as protein kinase B) seem to make the islet β-cells activation by regulation of the key inflammatory cytokines [8], alterations in the PI3K/AKT signaling pathway may contribute to specific therapeutic effects for diabetes. In addition, the physiological function of PTEN is to dephosphorylate the second messengers generated by the PI3K, thereby down-regulating insulin signaling downstream of the PI3K [9]. Therefore, potential role of these PI3K/AKT/PTEN signaling seems to be involved in the development of diabetes. For example, expression of constitutively active AKT linked to an insulin gene promoter in transgenic mice results in increased islet β-cells mass by altering β-cells size and number [10]. These mice are also resistant to diabetes [10]. Thus, the signaling activation may be of importance in both islet β-cells proliferation and survival. Searching for agents to promote pancreatic β-cells survival and improve its function could be a promising strategy to prevent and to treat for diabetes. Here, we provide an overview of research on the characterization of the PI3K/AKT/PTEN pathway at the viewpoint of pathogenesis for diabetes. Overall, certain diets and lifestyle as well as the synthesis of serotonin may be the most helpful in preventing type-2 diabetes, which are all involved in the PI3K/AKT/PTEN signaling pathway. Intervention and therapy that alter the pathogenesis may serve to reduce the risk of the development of the disease, leading to better efficacy of therapeutic approaches.
2. CELLULAR FUNCTION OF THE PI3K/AKT PATHWAY IN DISEASES
PI3Ks are crucial signaling components in a diverse range of cellular functions including cell proliferation, migration, and survival. It is reasonable that dysregulation of the pathway has been implicated in the induction and/or progression of a variety of diseases. The PI3K forms a family that can be divided into three classes based on the structure and mechanism of activation [11]. These PI3Ks are heterodimers consisting of a regulatory subunit (p85) and a catalytic subunit (p110). AKT is a major downstream target of those PI3Ks. Human AKT has three isoforms, AKT1, AKT2, and AKT3 [12]. A product of PI3K, PIP3, leads to the membrane recruitment of AKTs, and also binds to phosphoinositide-dependent kinase 1 (PDK1) via the plekstrin homology (PH) domains, then, PDK1 phospho-rylates AKT in the kinase domain (Thr-308 in AKT1). For the further activation of AKT, the phosphorylation within the carboxyl site (Ser-473 in human AKT1) by PDK2 is required [13]. After activated, AKT moves to the cytoplasm and nucleus. Then, AKT phosphorylates many downstream targets to regulate various cellular functions (Fig. 1). AKT substrates include GSK3β, Bad, Caspase-9, IKKα, NOS, TSC2, p27, MDM2, and so forth [14]. AKT mediated phosphorylation of these target proteins leads to their activation or inhibition. For example, AKT inhibits the GTPase-activating protein (GAP) activity of the tuberous sclerosis complex 1 (TSC1) and TSC2 by phosphorylating TSC2, leading to the accumulation and activation of mTOR [15]. The other AKT substrate, GSK3β, is also a serine/threonine kinase that was initially identified as playing a role in the regulation of glycogen synthesis in response to insulin receptor stimulation [16]. The S6 ribosomal protein belongs to the PI3K/AKT/PTEN/mTOR signaling pathway, and is phosphorylated by p70S6 kinase [17]. It has been shown that a negative feedback loop runs from p70S6 kinase to the upstream insulin receptor substrate-1 (IRS1) through the PI3K/PDK1/AKT pathway, suggesting a mechanism for the development of insulin resistance [18]. The IRS1 associated PI3K/AKT activity is severely impaired in high fat-fed animals [19]. In addition, the PI3K/AKT activation by insulin is associated with translocation of glucose transporter type 4 (GLUT4) [20]. Altered activity of these mediators then causes various effects on several cytokine expressions. Consequently, activation of PI3K/AKT pathway results in the inhibition of pro-inflammatory events such as expression of type-I interferon, IL-12, and TNFα, whereas the mTOR seem to up-regulate the anti-inflammatory cytokines [21].
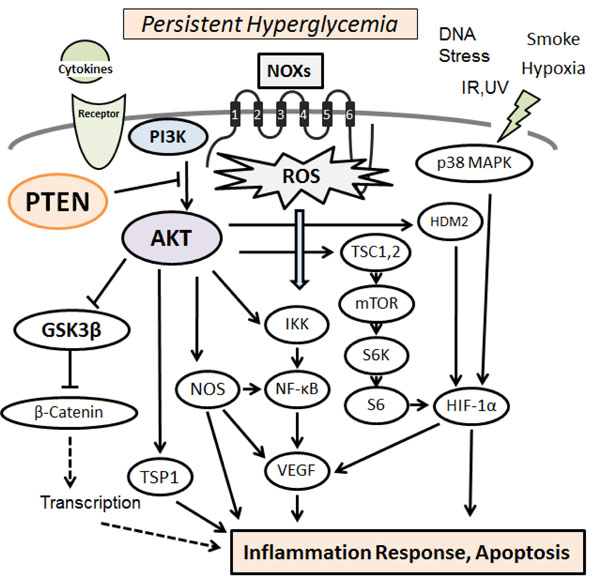
Hypothetical representation of PI3K/AKT/PTEN signaling in pancreatic β-cells is shown, which modulate NOXs-derived reactive oxygen species (ROS) generation. The NOXs sense persistent hyperglycemia, DNA stress, hypoxia, and so on. Uncontrolled generation of ROS might then contribute to the various pathway signaling. Examples of molecules known to act on the regulatory pathway are shown. Arrowhead means stimulation whereas hammerhead represents inhibition. Note that some critical pathways have been omitted for clarity.
The structure endows PTEN with its preference for phospholipid substrates such as PIP3. PTEN is a dual-specificity phosphatase which has protein phosphatase activity and lipid phosphatase activity that antagonizes the PI3K activity [22], acting as regulator of keeping basal levels of PIP3 below a threshold for the signaling activation. Under the basal conditions, enhanced insulin-PI3K signaling via deletion of PTEN leads to increased pancreatic β-cell mass because of both increased proliferation and reduced apoptosis [23]. In particular, the relationship between PTEN function and adipocyte-specific fatty-acid-binding protein FABP4 is of interest in β-cell signaling [24]. The interaction of PTEN to FABP4 suggests a role for this phosphatase in the regulation of lipid metabolism [25]. In PTEN-deficient mice, there is increased synthesis and storage of triglyceride due to the up-regulation of PI3K/AKT activity [26]. Hence, PTEN exerts a critical negative effect upon islet β-cell function. PTEN may be an important negative regulator of insulin-stimulated glucose uptake [27]. However, tissue targeted deletion of PTEN lead to improved insulin sensitivity in the insulin-responsive tissues, and protects from diabetes [23, 28]. In addition, even the partial reduction of PTEN is enough to elicit enhanced insulin sensitivity and glucose tolerance. PTEN has been shown to be up-regulated in insulin resistance model of insulin or insulin-like growth factor-1 signaling ablation in β-cells [29]. Furthermore, PTEN expression in pancreatic islets is up-regulated in models of type-2 diabetes [30]. Accordingly, controlling PTEN expression could be a therapeutic target to prevent the degeneration of β-cells. It has been shown that PTEN activity can be regulated by the post-translational regulation including phosphorylation, acetylation, and oxidation [31].
3. OXIDATIVE STRESS DAMAGES ISLET-CELLS VIA THE PI3K/AKT PATHWAY
Type-2 diabetes is characterized by diminished pancreatic β-cells function that is to secrete insulin in response to changes at extracellular glucose concentration [32]. Insulin signaling within the β-cells has also been shown to play an important role in maintaining the function of the β-cells. Defects in insulin secretion and decreased sensitivity of target tissues to insulin action are the key features of type-2 diabetes [32, 33]. Evidences indicate that mitochondrial dysfunction is a contributor to β-cells failure in the development of diabetes. Increased intra-cellular generation of ROS via the mitochondria seems to be implicated in the pathology of diabetes [34]. Hence, interventions that attenuate ROS-influences on β-cells might prevent or slow down the development of diabetes. Oxidative stress occurs as a result of an imbalance due to excess ROS production over the capability of the cells to support effective antioxidant responses, which can cause various metabolic complications including pancreatic β-cells damage. In particular, NADPH oxidases (NOXs) are the principal source for cellular ROS as a result of metabolic stresses [34, 35]. Several stress-response pathways including ROS leads to reduced ATP content and synthesis in β-cells, which is a critical parameter in regulating glucose-stimulated insulin secretion [36]. Moreover, ROS oxidize polyunsaturated fatty acids in mitochondrial cardiolipin and other phospholipids [37], which impair membrane integrity and leads to cytochrome-c release into cytosol and consequent cell apoptosis. Persistent hyperglycemia during diabetes also exerts toxic effects on islet cells by creating redox imbalance arising from overproduction of the ROS [38]. Accumulation of ROS disturbs the integrity and physiological function of cellular biomolecules impairing functionality and viability of the islet cells [39]. The ROS are also produced in cellular response to inflammatory cytokines and to bacterial invasion [40, 41], whose signal activates a chains of stress pathways involving a family of serine/threonine kinases including AKT and/or p38 MAPK (Fig. 1), which in turn have a negative effect on insulin signaling [42]. Animals with diabetes shows histological changes including inflammation foci, increased oxidative damage, up-regulated levels of inflammatory cytokines, and apoptotic cells in islet β-cells. In addition, ROS directly interact with critical molecules including PI3K, AKT, PTEN, and several phosphatases to initiate signaling in a variety of cellular processes such as proliferation and survival [43]. Data suggest a correlation between insulin sensitivity/insulin resistance and ROS levels [44, 45]. Consequently, the excessive oxidative environment may contribute to mitochondrial dysfunction, β-cells dysfunction, and impaired glucose tolerance, which can lead to the diabetic disease.
Activation of NF-kB by IKK occurs in association with ROS produced by NOXs in response to activation of IL-1β and/or TNFα receptors [46]. AKT and NF-kB inducing kinase (NIK) are involved in these processes [47]. ROS activate PI3K and inactivate PTEN by oxidizing cysteine residues within the active center [48]. AKT is also activated by inactivating protein Ser/Thr phosphatases via the ROS [49]. In addition, ROS could play a critical role in β-cells dysfunction through PTEN-dependent JNK activation and AKT inhibition [50]. On the other hand, serum withdrawal kills human U937 cells by elevating cellular ROS levels, which occurs through PI3K activation [51]. LY294002, a PI3K inhibitor, has been shown to reduce chemokine-induced ROS generation [52]. Since wortmannin, another PI3K inhibitor, also inhibits the translocation of NOXs subunits and reduces ROS production, in which the role of PI3K signaling might be involved [53, 54]. It has been reported that cell membrane depolarization, which is followed by PI3K/AKT and PKC activation causes NOXs assembly and ROS production. The ROS production is abolished by the wortmannin as well as the PKC inhibitor H7 [55], indicating that PI3K/AKT and PKC are in the same pathway and are required for the ROS production. In this way, PI3K/AKT pathway plays an apparent role in various oxidative stress pathways (Fig. 1). Oxidative stress is also implicated in the pathogenesis of free fatty acids (FFAs)-induced β-cells dysfunction [56]. The combination of obesity and type-2 diabetes is associated with elevated plasma FFAs which lead to apoptosis of β-cells through PTEN-dependent AKT inhibition, followed by reduced insulin expression and secretion [57].
4. SEROTONIN PROTECTS ISLET-CELLS VIA THE PI3K/AKT PATHWAY
Pancreatic islet cells share common developmental features with serotonin-producing neurons [58]. Serotonin reuptake transporter deficient mice exhibits glucose intolerance and insulin resistance and progressively developed obesity, which exhibited beta-cell hyperplasia, islet-mass expansion, and hyperinsulinemia [59]. Accordingly, it is probable that serotonin is one of the restorative factors for islet β-cells. Serotonin (5-hydroxytryptamine), an well-characterized neurotransmitter in the central nervous system, plays a crucial role in regulating mood, sleep, appetite, body temperature, and metabolism [60]. Some of the serotonin receptors, which are generally considered to be localized in the nervous systems, are also involved in processes associated with gastro-intestinal functions including the secretion and motility [61]. Actually, most of the serotonin is synthesized and localized in the gastro-intestinal tract, particularly in the entero-chromaffin cells [62]. Serotonin also regulates glucose-stimulated insulin secretion from pancreatic β-cells [63]. In addition, selective serotonin reuptake inhibitors (SSRIs) inhibit insulin secretion in pancreatic β-cells [64]. The islet β-cells proliferate during pregnancy in adaptation to the insulin resistance, which is accompanied by an increase of plasma serotonin level. The receptors mediating the action of serotonin are mainly classified into 7 major groups known as serotonin receptors [65]. The 5-HT-3 receptor is distinguished from among the other serotonin receptor subtypes because it is an associated ion channel, whereas the other subtypes serve as G-protein-coupled receptors [66]. Findings have provided evidence of the role of serotonin receptors in the regulation of inflammatory responses [67]. In fact, several serotonin receptor antagonists improve intestinal inflammation [68].
Studies have indicated that serotonin exerts part of their actions by modulating the activity of PI3K/AKT pathway [69]. Through the G-protein coupled receptors, serotonin activates the PI3K/AKT/mTOR/p70S6K signaling (Fig. 2), and this activation is similar to that seen in VEGF signaling [70]. One of the major functions of the PI3K/AKT pathway is to promote growth factor-mediated cell survival and to block cell apoptosis. GSK3β has a role for the regulation of serotonin receptor in cell surface trafficking [71]. Antidepressants acting on serotonin neurotransmission activate AKT and inhibit GSK3β. Lithium also activates PI3K/AKT signaling. In addition, drugs like SSRIs and MAO inhibitors that elevate serotonin synaptic transmission have been shown to inhibit GSK3β [72]. The mood stabilizers such as valproate also inhibit GSK3β [73]. Defective central AKT function alters serotonin signaling as well as serotonin associated behaviors, demonstrating a novel role for PI3K/AKT pathway in maintaining serotonin receptor function [74]. It is plausible that PI3K/AKT/GSK3β pathway also functions downstream the serotonin receptor in pancreatic islet β-cells.
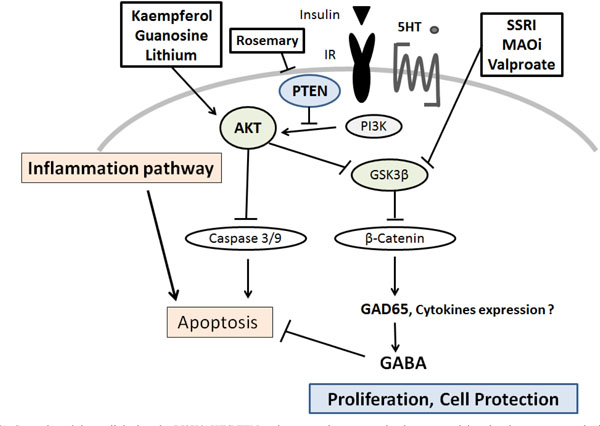
Several modulators linked to the PI3K/AKT/PTEN pathway are demonstrated, whose potential molecular targets may be based on the predominant sites. Arrowhead means stimulation whereas hammerhead represents inhibition, suggesting implication of PI3K/AKT/ PTEN modulators for the therapy of diabetes via the pancreatic β-cells protection. Note that some critical events have been omitted for clarity.
SSRI: Selective Serotonin Reuptake Inhibitors
MAOi: Monoamine oxidase inhibitors
5-HT: 5-hydroxytryptamine, serotonin
Gamma-aminobutyric acid (GABA) has been shown to inhibit apoptosis of animal β-cells [75] (Fig. 2). GABA activates three types of membrane receptors. It has been shown that β-cells express the GABA receptors and produce GABA through GAD65 [76]. The GAD65 is a crucial target of auto-anti-GAD65 antibodies associated with the development of type-1 diabetes [77, 78]. Augmentation of the GAD65 expression is induced by serotonin treatment, in which the phosphorylation of AKT is activated, and specifically repressed by inhibitors of PI3K such as LY294002 [79]. Activation of the GABA receptors in islet β-cells increases insulin release, exerts protective effects on islet β-cells, and reduces cell apoptosis in islet cells [80, 81]. GABA receptor also acts as a chemo-attractant receptor via an AKT-dependent pathway in neutrophils [82]. In addition, GABA has been shown to increase DNA synthesis in pancreatic cell line [81]. Therefore, GABA increases β-cells proliferation, protects islet cells from apoptosis, and prevents hyperglycemia [83]. Although there are clear physiological connections behind the effects, the clinical relevance needs to be studied further.
5. CERTAIN DIET AND LIFESTYLE MAY CONTRIBUTE TO ISLET Β-CELLS PROTECTION
Some of anti-inflammation drugs and antioxidants function as a blocker of radical processes by eliminating harmful ROS produced during normal cellular metabolism [84, 81]. In addition, several antioxidants could be used as a supplement to scavenge local ROS thereby improving the survival of islet β-cells, because their survival may be crucial for achieving successful therapy [85]. Realizing the susceptibility of islet β-cells to oxidative damage, several efforts to enhance their defense mechanism in the form of oral administration of antioxidants have proven successful [86]. Besides, oxidative damage can be reduced by controlling hyperglycemia, calorie intake, minerals, and vitamins [87]. At present, promising regulators for PI3K/AKT pathway for the islet β-cells survival may be as follows. Guanosine can protect cells from mitochondrial oxidative stress by activating the PI3K/AKT/GSK3β signaling and by induction of the antioxidant enzyme HO-1[88]. Guanosine is a purine nucleoside that is found in RNA, which may be produced from dried fish or seaweed and could be used as food additives for flavor enhancers. Melatonin also prevents cell-injury expansion through the PI3K/AKT dependent HO-1 expression [89], which is a hormone produced by the pineal gland. Activation of PI3K/AKT/GSK3β signaling may not only up-regulate the HO-1 expression, but also the protective effects of this pathway may be linked to the actual effects of HO-1. Melatonin is found in many common fruits and vegetables including grape skins, tomatoes, and walnuts. In addition, there are numerous foods such as bananas, pineapples, and oranges to naturally boost melatonin production [90]. It is important to exploit the potential benefits of these agents for diabetes. Several herbs may also be promising. Kaempferol is a flavonol that is present in various plants including grapefruit and some edible berries. Kaempferol treatments promote viability, inhibited apoptosis, and reduced caspase-3 activity in human islet β-cells. In addition, Kaempferol also induces the activation of PI3K and AKT [91]. Kaempferol treatment improve the expression of anti-apoptotic AKT that is significantly reduced in islet β-cells chronically exposed to hyper-glycemia [92]. Epoxypukalide, a furanocembranoid from gorgonian leptogorgia setacea, induces β-cells proliferation and protects against cytokine-mediated β-cells death in cultured islet cells [93]. Interestingly, rosemary (Rosmarinus officinalis) exerts remarkable anti-diabetogenic effect [94]. Some of rosemary extract represses PTEN expression in K562 leukemic culture cells [95].
Diet promotes sleep duration and quality, which is an essential part of life [96]. Epidemiologic studies have demonstrated that those who sleep less are more probable to eat energy-rich foods including fats and carbohydrates [96]. Furthermore, increasing evidence shows that sleep has an influence on dietary choices [96]. As serotonin secretion may occur in a good situation of sleep, mood, body temperature, appetite, and metabolism and so forth, these conditions should be well-maintained to prevent diabetes and to avoid the disease progression by proper serotonin secretion. Interestingly, treatment of ghrelin, a hormone that stimulates hunger, restores PI3K/AKT pathways [97]. On the other hand, overexpression of PTEN has been shown to have inhibitory effects on insulin signaling, including decreased AKT activity and GLUT4 translocation to the cell membrane, showing the contribution to insulin resistance [98, 99]. In contrast, down-regulation of PTEN has the opposite effect with increased glucose uptake in response to insulin [100]. Inhibiting the signaling via PI3K/AKT, PTEN indirectly promotes serotonin synthesis and secretion [101]. There is crosstalk between PTEN and serotonin receptor [102]. Honokiol, a chemical compounds in traditional eastern herbal medicines by the way, can attenuate the PI3K/AKT signaling by up-regulation of PTEN expression [103]. Combination of honokiol with the mTOR inhibitor rapamycin presents the synergistic effects [104]. In addition, molecular targets including phosphorylated AKT and mTOR are down-regulated by the combination of curcumin and resveratrol due to the activation of PTEN [105]. These findings might be translated into new dietary treatments for diabetes in the future.
6. PERSPECTIVE
Several non-insulin anti-hyperglycemic drugs have been approved for the treatment of type-2 diabetes mellitus, which largely comprise three groups by the pharmacological targets: glucose balance, insulin secretion, and metabolic improvement (Fig. 3). The representative drugs are α-glucosidase inhibitors, sulfonylureas and the dipeptidyl peptidase-4(DPP-4) inhibitors, biguanides and dual peroxisome proliferator-activated receptors (PPAR) agonists (thiazolidinediones or glitazones). Diets and some serotonin therapy presented here may be involved in the insulin secretion group through the effect of the islet β-cells protection (Fig. 3). Now, there is a progression from monotherapy to multiple-therapy with or without additional agents in the treatment of diabetes. However, for example, anti-diabetic drugs often present proven or potential cardiac hazards [106]. So, optimal use of treatments for diabetes is of importance to the sustainability of health care. The precise involvement of the PI3K/AKT/PTEN /GSK3β/mTOR in islet β-cells signaling has remained unexplored. Further research is therefore required to determine whether agents differ in terms of long-term complications of diabetes and mortality. In addition, the cost-effectiveness of these drugs requires further study. More understanding of the precise intracellular mechanisms and variations in pathogenesis of diabetes could provide novel insights into the development of therapeutic approaches against diabetes.
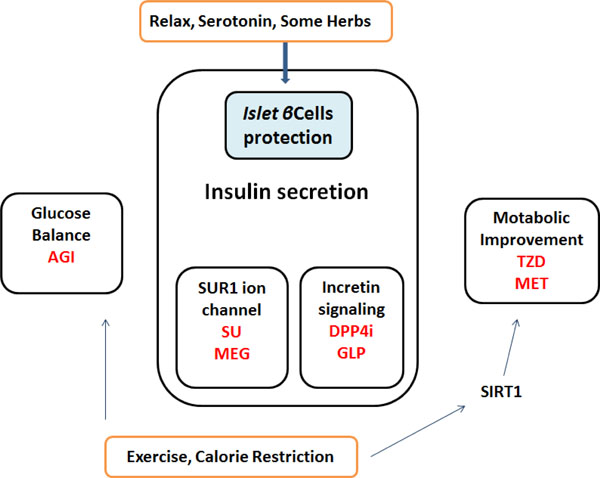
Simplified diagrams indicating the medicinal properties of non-insulin diabetes-treatments are shown. Representative drugs in a group are also presented. Note that some critical drugs have been omitted for clarity.
Annotated abbreviations used here;
AGI: α-Glucosidase inhibitors
DPP4i: Dipeptidyl-peptidase-4 inhibitors
GLP: Glucagon-like peptide-1 agonists
MEG: Meglitinides
MET: Metformin
SU: Sulfonylurea
TZD: Thiazolidinedione
SIRT1: a member of the sirtuins, a distinct class of trichostatin A-insensitive lysyl-deacetylases
ABBREVIATIONS
CONFLICT OF INTEREST
The authors declare that they have no competing financial interests.
ACKNOWLEDGEMENTS
This work was supported by JSPS KAKENHI Grant Number 25560050, 26-12035, 24240098.

