All published articles of this journal are available on ScienceDirect.

NO Metabolites Levels in Human Red Blood Cells are Affected by Palytoxin, an Inhibitor of Na+/K+-ATPase Pump
Authors Info & Affiliations
Abstract
Palytoxin (PTX), a marine toxin, represents an increasing hazard for human health. Despite its high toxicity for biological systems, the mechanisms triggered by PTX, are not well understood. The high affinity of PTX for erythrocyte Na+/K+-ATPase pump is largely known, and it indicates PTX as a sensitive tool to characterize the signal transducer role for Na+/K+-ATPase pump. Previously, it has been reported that in red blood cells (RBC), probably via a signal transduction generated by the formation of a PTX-Na+/K+-ATPase complex, PTX alters band 3 functions and glucose metabolism. The present study addresses the question of which other signaling pathways are regulated by Na+/K+-ATPase in RBC. Here it has been evidenced that PTX following its interaction with Na+/K+-ATPase pump, alters RBC morphology and this event is correlated to decreases by 30% in nitrites and nitrates levels, known as markers of plasma membrane eNOS activity. Orthovanadate (OV), an antagonist of PTX binding to Na+/K+-ATPase pump, was able to reverse the effects elicited by PTX. Finally, current investigation firstly suggests that Na+/K+-ATPase pump, following its interaction with PTX, triggers a signal transduction involved in NO metabolism regulation.
INTRODUCTION
Palytoxin (PTX), a marine toxin (Fig. 1) isolated from the Cnidaria (zooanthid) and Ostreopsisdinoflagellates, represents one of the most well-known potent non-protein substances [1]. It is a very complex molecule that presents both lipophilic and hydrophilic parts. In the last years, palytoxin (PTX)-producing Ostreopsis have been identified along the coasts in Mediterranean area [2,3]. Extremely low concentrations of PTX, following its binding to Na+/K+ ATPase, is able to depolarize the cell by inducing a lower conductance and the appearance of relatively non-selective cation channels which likely include, at least, part of the pump’s ion translocation pathway [4-7]. Altered ionic homeostasis, induced by PTX, influences other equilibria such as those of Ca2+ and H+ ions, inducing a collapse of the mechanisms controlling the ion-water balance, resulting ultimately for several cellular effects [5,8,9]. Na+/K+ ATPase, as shown by data obtained with oubain, represents the main binding site for PTX [10]. On the other hand, vanadate, a specific inhibitor of all type-P ATPases (which the Na+/K+-ATPase belongs), inhibits PTX mediated effects acting like an analogue of phosphate [7, 11], which is able to form a stable complex with Na+/K+-ATPase on the inner side of the red blood cell (RBC) membrane, stabilizing the dephosphorylated form [12]. It has been hypothesized that PTX exerts its toxicity by modulating actin filaments within cytoskeleton proteins [13]. Studies on neuronal cells of human neuroblastoma BE (2)-M17, performed in order to evaluate the anti-cytoskeletal effects and the cationic flux changes induced by the marine toxins, showed that PTX triggers a series of cytotoxic events which range from the rapid depolarization and cytosolic Ca2+ increase to the cyto-architecture reorganization [14]. In rather recent studies it has been suggested that Na+/K+-ATPase, as a result of its interaction with PTX, may act as a signal transducer, activating some mechanisms which are closely associated with the cytoskeleton [15,16].
Chemical structure of Palytoxin.
RBCs can be considered as oxygen sensor, in fact when they pass through the microcirculation they may sense tissue oxygen conditions via their deoxygenation degree and couple this information to the vasodilatory compounds release, such as ATP or nitric oxide (NO), that enhance blood flow to hypoxic tissues [17]. Furthermore, NO has been hypothesized to regulate RBC deformability by modulating physiochemical properties of the cytoskeleton [18] and a functional endothelial type nitric oxide synthase (eNOS) located in plasma membrane has been reported to be expressed in RBC [19]. It is also known that impaired Na+/K+-ATPase activity may contribute to the decrease of RBC deformability, due to intracellular calcium increase, resulting ultimately in altered membrane skeleton interactions [20]. PTX, as a consequence of its high affinity for erythrocyte Na+/K+-ATPase pump [21], is a sensitive tool for studying in RBC the relationship between Na+/K+-ATPase pump, morphological alterations, NO metabolism and ATP release from RBC.
The aim of this study is to achieve a significant step toward elucidating the mechanisms by which PTX-mediated signals are transduced through cells. In this study, we investigate the potential coupling of the reactions of RBCs with PTX, nitrite metabolism and the release of ATP from RBCs. By demonstrating that PTX inhibits the release of ATP from RBC by a mechanism NO dependent, a new potential mechanism for PTX-mediated toxicity is presented.
MATERIALS AND METHODS
Chemicals
All reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA). Blood samples, collected in citrate, were obtained from ten informed healthy donors (aged 30±5 years), excluding blood donors, who declared that they had abstained from all drug treatments for at least one week prior to sample collection, in accordance with the principles outlined in the Declaration of Helsinki.
Preparation of RBCs
Healthy human blood samples were washed by centrifugation with the incubation buffer (Hepes 25 mM, NaH2PO4 1 mM, NaCl 110 mM, KCl 5 mM and 2 mM MgCl2, at pH 7.4 and 290 ± 5 mOsm•Kg-1) with the purpose to separate and isolate red blood cells. The same buffer was used to re-suspend RBCs to 3% hematocrit.
Methemoglobin (met-Hb) levels and the degree of hemolysis were determined at the end of the incubation time according to Ziijlstra et al. [22].
Measurement of Intra-extracellular ATP
ATP concentration was measured at both intracellular and extra cellular levels as previously described [23] using the luciferin-luciferase technique. This method quantizes the amount of light generated in the reaction between the ATP and the firefly tail extract, which is dependent on ATP concentration. The light emitted was quantified by the 1251 luminometer BioOrbit.
Measurements were conducted on erythrocytes after 30 minutes of incubation at 37°C, with PTX (1 pM) and OV (1mM). Treated RBCs were diluted in a ratio 1:100 and incubated for 20 min. with the direct activator of Gi, Mastoparan 7 (4 µM Mas 7). To measure the total intracellular ATP, all the ATP consuming processes were blocked with trichloroacetic acid (TCA) which was diluted prior to testing, to avoid interferences. Values were normalized to ATP concentration per erythrocyte.
Preparation of Red Blood Cells and Microscopy
After blood collection, plasma separation was obtained by centrifuging at 2500g for 5 min. Red blood cells were isolated by density gradient centrifugation by Ficoll. After separation procedure, the packed cells were gently re-suspended with the buffer, reported previously (Hct 10%). PTX concentration was fixed to 1 pM, as reported previously [24]. To exclude the possibility that erythrocyte lysis could alter results after treatment, red blood cells were sedimented by centrifugation at 500g for 10min. The presence of 69aemoglobin in the supernatant was determined by light absorption at 405 nm (Cary 3E, Varian, Palo Alto, CA). Cell morphology was analysed by a Nikon microscope (40X objective), where images were collected by using a digital Nikon camera (Coolpix 5400) with the X-Pro software.
Measurement of Nitrite and Nitrate Formation as Markers of eNOS Activity
Cells were incubated in the above incubation buffer at 37 °C, in the absence or presence of 1.0 pM of PTX, 1.0 mM of orthovanadate (OV), under different experimental conditions. As a consequence of their stability, nitrites and nitrates are widely known to be markers for NO production in biological systems [25]. Measurements were executed according to the Griess Reagent Kit instructions (Sigma-Aldrich, St. Louis, MO, USA).
Statistical Analysis
Data are presented as mean ± standard deviation (S.D.). Statistical calculations were performed with the software Excel (Microsoft, CA, USA). A Wilcoxon test was used to analyze the differences of measured parameters before and after treatment. Since different erythrocyte specimens used in distinct experiments have different susceptibility to treatments, therefore, the same erythrocyte specimens have been used for control and experimental conditions. Statistical significance was set at P ≤ 0.05.
RESULTS
PTX and RBC Morphology
When human red blood cells were incubated with PTX, cell morphology was monitored by means of a phase-contrast microscopic observation. In Fig. (2B), after 2-h treatment with 1 pM PTX, a concentration routinely used to study PTX mediated-toxic effects on [26] treated cells showed a slightly altered morphology with irregular shapes, suggesting the presence of cytoskeleton arrangements induced by PTX. By contrast, as shown in Fig. (2A), in control cells any significant morphological alteration was not evident.
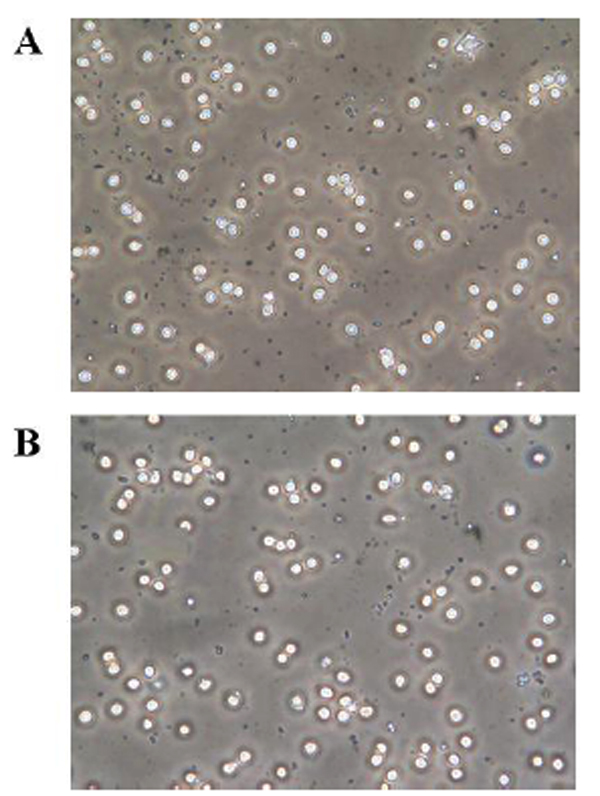
Effects of palytoxin (PTX) on erythrocyte morphology. Red blood cells were incubated with medium alone (A) and with PTX (B) for 2 h. Experimental procedures are described in Materials and Methods.
Effects of PTX on ATP Release from RBC
To support other evidences relating to mechanical stimulation of the toxin on the RBC, we evaluated its influence on ATP release from RBC comparing the effect to the orthovanadate one (OV), a molecule that inhibits the PTX inhibitory action acting as an analogue of phosphate on Na+/K+ ATPase and Mas 7 a direct activator of the heterotrimeric G protein [27]. The results showed in Fig. (3A) clearly indicate that RBC pretreated with PTX for 2 h released significantly more ATP than RBC in normal conditions; similar to what was observed in the presence of Mas 7. Differently, RBC pretreatment with OV do not show any significant effect on ATP release. These results highlight a link between the ATP release from the RBC and interaction of the toxin with the Na+/K+ ATPase. In fact, OV seems to be able to stop the signal transduction triggered by the PTX that leads to the release of ATP. Moreover, since OV is known to affect protein phosphorylation, including cytoplasmic tail of band 3 [28], we can assert that the release of ATP is not connected with the phosphorylation state of the RBC. The increase in ATP release induced by PTX, was accompanied by a reduction in cytosolic ATP content of the red blood cells (Fig. 3B), probably due to the increase in H+ concentration following the toxin binding to Na+/K+-ATPase. Therefore, as shown in Fig. (3B), OV induces a stronger decrease in intracellular ATP with respect to that shown by PTX.
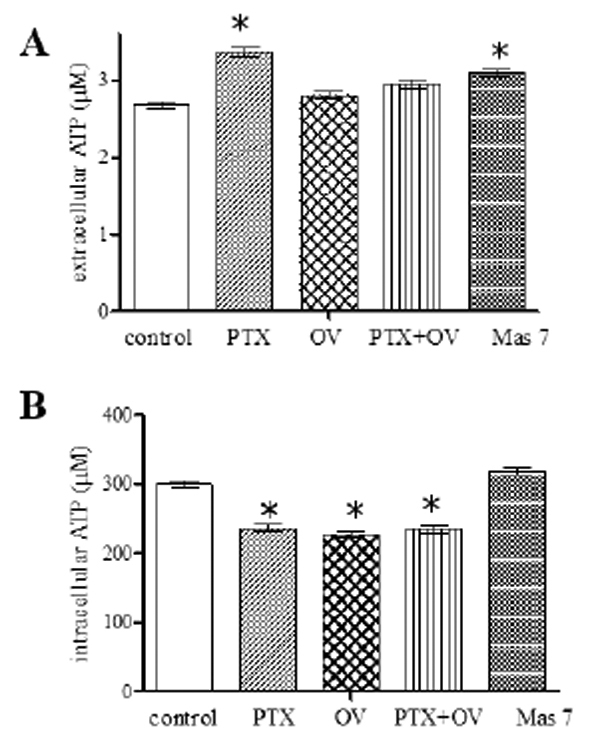
Extracellular (A) and intracellular (B) ATP levels measured at the end of the incubation time of red blood cells without (control) and with respectively 1 pM palytoxin (PTX), 1mM orthovanadate (OV), 1 pM PTX plus 1mM OV, 4 μM Mas 7. Results are from four independent experiments ± SD. P<0.05 vs. Starting point.
Effect of PTX on Nitrites and Nitrates Production in RBC Suspensions
NO has been hypothesized to regulate erythrocyte deformability [18]. On these basis, in order to test whether PTX-mediated cytoskeleton alterations may have negative effects on membrane eNOS, nitrite and nitrate levels were measured in treated and untreated red blood cells as markers of NO production [20]. The exposure of human red blood cells to PTX alters nitrite and nitrate levels in the cell. As shown in Fig. (4A), PTX treatment reduces nitrate and nitrite production in a time dependent manner. The maximum effect was observed after 24 h of incubation but it was already significant after 2 h. The inhibitory effect shown by PTX at 2 h on nitrite and nitrate production (Fig. 4B) was partially recovered by OV. OV alone did not show any significant effect on nitrate and nitrite levels.
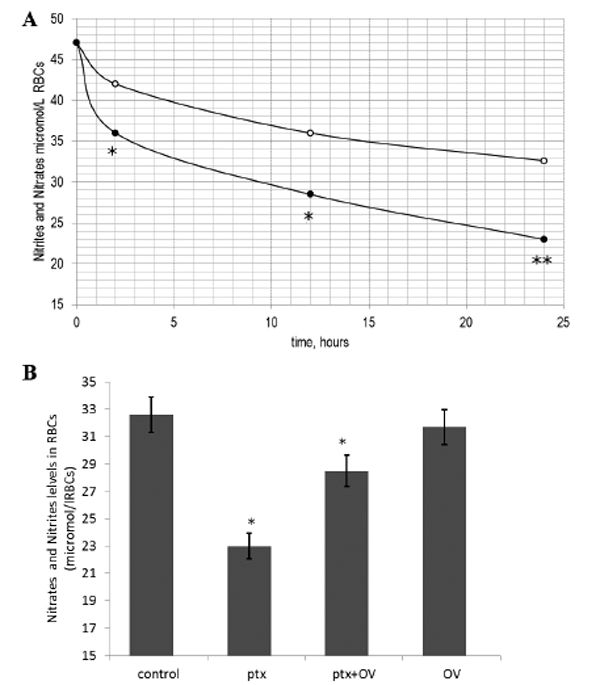
Time course of the effects of palytoxin (PTX) on nitrite and nitrate levels (A). Cells were incubated with medium alone (o) or with medium containing PTX (•). Results are from three independent experiments. * P < 0.05 vs. control; (B) Effects of PTX on nitrite and nitrate levels in red blood cells in the presence and absence of orthovanadate 1mM (OV) after 2h. Results are from three independent experiments. * P < 0.05; **P<0.001 vs. control.
Haemolysis Degree and MetHb Levels
An obvious potential source of extra-cellular ATP, nitrite and nitrates is the spontaneous erythrocyte lysis. To determine cell lysis after experiments, red blood cells suspensions were analysed to evaluate haemoglobin concentration in the supernatant. The percentage of hemolysis was always less than 1.5%. MetHb levels determined in intact red cells after treatments was, respectively, 3.3±2.3 and control cells 2.6±4.5%, N=5, P = 0.3.
DISCUSSION
Previous results showed that red blood cells (RBC) treated with PTX showed a strong decrease of the anion transport i.e. band 3 [26], suggesting that this molecule which is largely known to induce its toxicity through Na+/K+-ATPase inhibition is able to affect indirectly other essential RBC structures like band 3, probably via a signal transduction generated by the formation of a PTX-Na+/K+-ATPase complex. The present study extends this previous observation [26], by demonstrating that PTX, following its interaction with Na+/K+-ATPase, induces a strong decrease in NO derived metabolites levels (i.e. nitrites and nitrates), that are widely used markers for NO production in biological systems [19,29]. Recent studies demonstrated that human red blood cells have an active and functional endothelial type NOS, which is located within the plasma membrane [19]. It is known that PTX appears to exert its effect by modulating the cytoskeleton properties, in particular by acting on actin filaments [13]. Thus, PTX, following the binding to Na+/K+-ATPase pump, could affect cytoskeletal rearrangement, resulting in NO production decrease by affecting function of eNOS sited on inner leaflet of plasma membrane that is known to be able to regulate deformability and death [19,30] of RBCs. On this line, we observed microscope, the slight RBC morphological changes, that are correlating with cytoskeletal rearrangements, occurred following PTX short exposure. Further studies should be performed to clarify the contribution of thiol groups, i.e. GSH and Hb, in the observed alteration of NO bioavailability, following the PTX exposure [31, 32]. A higher PTX-induced harmful effect on RBC morphology is probably counteracted by PPP stimulation, reported previously [26].
Furthermore, NO is known to be an inhibitor of ATP release, via inactivation of the heterotrimeric G protein G(i) [33]. This finding offers a possible explanation regarding the mechanism underlying the observed increase in ATP efflux from RBC, following PTX exposure. It has been shown that RBC participate in the regulation of vascular resistance releasing ATP in response to physiological stimuli such as reduced oxygen tension and mechanical deformation [34]. Thus, the initiating signal involved in ATP release from RBC might involve in cytoskeleton conformational changes induced by the formation of the PTX-Na+/K+-ATPase complex, that causes stress on the membrane components, thus activating the downstream pathway relating to ATP release [35]. ATP released from RBC is involved in vascular functionality [36]. The mechanism of ATP release has been an area of intense investigation, but so far the problem is not fully resolved [37]. However, inhibition of PTX-induced ATP release by orthovanadate (OV) an antagonist of PTX on the Na+/ K+-ATPase [7], suggests that Na+/ K+-ATPase pump may be involved in such mechanism.
In conclusion, PTX-induced alteration on NO metabolism in vasculature, could represent, an event, involved in the mechanisms underlying PTX-mediated toxicity. Finally, this study ssfurther suggests that signaling function of the Na+/ K+-ATPase is pivotal to PTX-induced effects in RBC.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflicts of interest.
ACKNOWLEDGEMENTS
The financial support of University of Cassino and Southern Lazio (FAR 2012) to F.M. is gratefully acknowledged.

