All published articles of this journal are available on ScienceDirect.

High Density Lipoprotein and it’s Dysfunction
Abstract
Plasma high-density lipoprotein cholesterol(HDL-C) levels do not predict functionality and composition of high-density lipoprotein(HDL). Traditionally, keeping levels of low-density lipoprotein cholesterol(LDL-C) down and HDL-C up have been the goal of patients to prevent atherosclerosis that can lead to coronary vascular disease(CVD). People think about the HDL present in their cholesterol test, but not about its functional capability.
Up to 65% of cardiovascular death cannot be prevented by putative LDL-C lowering agents. It well explains the strong interest in HDL increasing strategies. However, recent studies have questioned the good in using drugs to increase level of HDL. While raising HDL is a theoretically attractive target, the optimal approach remains uncertain. The attention has turned to the quality, rather than the quantity, of HDL-C. An alternative to elevations in HDL involves strategies to enhance HDL functionality.
The situation poses an opportunity for clinical chemists to take the lead in the development and validation of such biomarkers. The best known function of HDL is the capacity to promote cellular cholesterol efflux from peripheral cells and deliver cholesterol to the liver for excretion, thereby playing a key role in reverse cholesterol transport (RCT). The functions of HDL that have recently attracted attention include anti-inflammatory and anti-oxidant activities. High antioxidant and anti-inflammatory activities of HDL are associated with protection from CVD.
This review addresses the current state of knowledge regarding assays of HDL functions and their relationship to CVD. HDL as a therapeutic target is the new frontier with huge potential for positive public health implications.
1. INTRODUCTION
1.1. Epidemiological Studies on HDL and its Protective Role in CVD
Epidemiological studies and prospective randomized trials have consistently shown a powerful inverse association between the magnitude of HDL-C and CVD [1]. In the Framingham Heart Study, each 0.26 mmol/L increase in HDL-C was associated with a significant decrease in relative risk for CVD mortality. Furthermore, the risk for restenosis following a vascular intervention is inversely related to HDL-C [1].
HDL has been shown to have a variety of functions that may contribute to its cardiovascular protective effects, including promotion of macrophage cholesterol efflux, RCT, anti-inflammatory and anti-oxidative effects.
1.2. HDL Quantity Versus HDL Quality
There are two different definitions for HDL; HDL quantity (circulating plasma levels of HDL-C) and HDL quality (atheroprotective properties of HDL). Plasma concentrations of HDL-C, while epidemiologically predictive of atherosclerotic cardiovascular events in large populations, are insufficient to capture the functional variation in HDL particles and the risk of CVD associated with HDL [2].
HDL-C is the cholesterol content within all of the α-HDL particles that exist in a deciliter of plasma. It makes no definitive statement about HDL particles (HDL-P), apoA-I or HDL size. HDL-P, determined by nuclear magnetic resonance spectroscopy refers to the number of HDL particles that exist in a liter of plasma [3]. Reliability of the sole measurement of plasma HDL-C has been questioned in the precise determination of CVD risk. Indeed, there are several genetic syndromes of very low HDL-C and apoA-I that are not associated with an increased risk of premature CVD.
So, measurement of HDL-C levels has no major relationship with how the HDL-P concentrations are being dynamically remodeled or the state of HDL functionality [4]. For example, deficiency of plasma LCAT does not appear to increase CVD risk despite HDL-C concentrations are <0.39 mmol/L. Even Tangier disease, caused by mutations in the ABCA1 transporter gene, in which HDL and apoA-I concentrations are virtually undetectable, is not associated with the marked increase in CVD that should be expected from such a dramatic phenotype [1].
2. HDL-STRUCTURE
HDL is highly heterogeneous, with subfractions that can be identified on the basis of density, size, charge, and protein composition. The concept that certain subfractions of HDL may be better predictors of CVD risk is of great concern [5].
HDL is the smallest of the lipoproteins particles with a size of 6-12.5 nanometers. HDL is composed of approximately: 55%protein, 3-15% triglycerides(TG), 26-46%phospholopids(PL), 15-30 cholesterol esters(CE), 2-10%choles-terol [6].
2.1. Major Proteins of the HDL
HDL constitutes a dynamic polydisperse group of particles which are central to lipid metabolism [7]. Its protein component is exceedingly diverse, comprising structural apolipoproteins, enzymes, co-factors for enzymes and numerous other proteins (Table 1). HDL proteins have traditionally been divided into 4 major subgroups: apolipoproteins, enzymes, lipid transfer proteins and minor proteins [8,9].
HDL Proteins Have Traditionally been Divided Into 4 Major Subgroups: Apolipoproteins, Enzymes, Lipid Transfer Proteins and Minor Proteins
| Apolipoproteins of the HDL Proteome | |||||||||||||
| ApoA-I | ApoA-II | ApoA-IV | ApoA-V | ApoC-II | ApoC-III | ApoC-IV | ApoD | ApoE | ApoF | ApoH | ApoJ | ApoL | ApoM |
| Enzymes of the HDL Proteome | |||||||||||||
| LCAT | PON1 | PAF-AH | GSPx | ||||||||||
| Lipid transfer proteins of the HDL proteome | |||||||||||||
| PLTP | CETP | ||||||||||||
The inhibition of LDL oxidation is a major antiatherogenic property of HDL [10]. This activity is due, in part, to HDL associated proteins. However, whether these proteins interact in the antioxidant activity of HDL is unknown. In an impressive study conducted to understand this situation; LDL was incubated with apolipoprotein A1 (apoA-1), lecithin:cholesterol acyltransferase (LCAT), and paraoxonase-1 (PON1) in the presence or absence of HDL under oxidizing conditions. When LDL lipid peroxide concentrations were determined, ApoA-1, LCAT, and PON1 were all found to inhibit LDL oxidation in the absence of HDL and enhance the ability of HDL to inhibit LDL oxidation [8,10].
The functional deficiency of HDL is intimately associated with changes in HDL composition. In addition to compositional changes to HDL that occur under pathological conditions, the protein or lipid components of HDL can be modified chemically [11,12]. Most humans have far more HDL particles than they do betalipoproteins (LDL and VLDL and IDL): HDLs are measured in micromolar concentrations where apoB particles are in nanomolar concentrations [3]. Each HDL particle carries apoAI and may also carry other apolipoproteins, such as apoAII, apoAIV, apoE, and apoC [13].
The most abundant protein in HDL, apoA-I, can be measured in plasma using widely available immunoassays [1]. Immunoaffinity chromatography has been used to classify HDL on the basis of apoA-I and apoA-II content. ApoA-I (molecular mass 28 kDa) is the major structural HDL apolipoprotein and accounts for 70% of total HDL protein, whereas the second major HDL apolipoprotein, apoA-II, represents the 20% [13].
ApoA-I is secreted as a lipid poor protein, and then rapidly acquires phospholipids and unesterifed cholesterol via ABCA1 transporter to form discoidal HDL particles. In most people, apoA-I is approximately one third and apoA-I:apoA-II is approximately two-thirds of the total HDL. Valuable metabolic differences have been reported between (A-I) HDL and (A-I/A-II) HDL [1,14].
Researchers have shown that the apoA-I in (A-I) HDL is cleared from normal human plasma more rapidly than the apoA-I in (A-I/A-II) HDL. This result could be interpreted in terms of the shorter plasma residence time of (A-I) HDL, causing an apparent enhancement of the cardio protective properties of (A-I/A-II) HDL [9,14].
2.1.1. ApoA-I and CVD
Apo A-I is synthesized in the intestines and liver and is thought to be largely responsible for the anti-atherogenic effects of HDL [15]. ApoA-I is also found on large TG-rich lipoproteins like chylomicrons. ApoA-I of mature HDL is catabolized by the liver, whereas lipid-poor apoA-I is catabolized by the kidneys [16, 17]. Multiple in vivo studies have demonstrated that apoA-I is a potent anti-oxidative and anti-inflammatory agent [8].
In addition, ApoA-I is a major player in RCT [18]. ApoA-I is a key component of HDL for cholesterol efflux, and its anti-atherogenic action has long been established [19]. ApoA-I attaches to cellular cholesterol efflux pumps (the major HDL lipidation pump or ABCA1) and is lipidated with free cholesterol and phospholipids: it is now referred to as prebeta-1 or ultimately prebeta-2 HDL [3]. This is the critical step in the formation of HDL particles. Without this lipidation step there will be no development of mature HDL [20].
Evidence is accumulating that apoA-I is the major anti-atherogenic and anti-oxidant factor in HDL. Whether plasma apoA-I concentrations are more predictive of CVD than HDL concentrations is somewhat nebulous that can be difficult to address statistically because of the very tight correlation between apoA-I and HDL. Recent data refer apoA-I concentrations to be superior to HDL-C in predicting CVD risk, but this approach has not yet been incorporated into national guidelines [21,22]. Furthermore, novel therapeutic interventions that increase apoA-I concentrations may be superior to those that primarily increase HDL concentrations, but this superiority remains largely theoretical at present [21]. Of interest, individuals heterozygous for the Milano mutation of the apoA-I gene do not develop early CVD despite HDL-C <20 mg/dL, and the situation is similar for the apoA-I Paris mutation [23].
The majority of cholesterol is transported as CE in lipoproteins including HDL [9]. ApoA-I carries an enzyme called LCAT which rapidly esterifies. Human HDL can also directly reduce cholesteryl ester hydroperoxides and phosphatidylcholine hydroperoxides via Met residues 112 and 148 of apoA-I [24]. This finding supports the key anti-oxidant role of apoA-I. In addition, apoA-I can remove oxidized phospholipids from oxidized LDL as well as from cells [25].
2.1.2. ApoA-II and CVD
The second most abundant protein in HDL is apoA-II. Clinical and epidemiological studies have yielded conflicting results regarding the relationship between plasma apoA-II levels and CVD: apoA-II is either pro-atherogenic or atheroprotective [26]. Nevertheless, apoA-II has long been considered to be of physiologically minor importance in lipoprotein metabolism because apoA-II deficiency is not associated with a high susceptibility to CVD [27].
HDL enriched in human apoA-II supports effective RCT from macrophages [28]. ABCA1 interacts with extracellular amphipathic apolipoproteins to initiate efflux of cholesterol from cells [29]. The −265C polymorphism in the apoA-II promoter region was shown to be associated with decreased plasma apoA-II concentration and enhance postprandial metabolism of large VLDL [30]. These results indicate that apoA-II plays an important role in both VLDL and HDL metabolism, possibly through the inhibition of LP activity. Presently, the exact roles of apoA-II, the second major HDL component, in the lipoprotein metabolism have not been fully understood [9].
2.1.3. Minor Apolipoproteins and Proteins of HDL
Minor HDL protein components (typically 10% of the HDL protein moiety) include apoE, apoA-IV, apoA-V, apoJ, apoC-I, apoC-II, and apoC-III, although the composition can vary markedly from person to person [8,9].
The third and fourth most abundant HDL apolipoproteins are apoA-IV and apoE respectively [9,31]. HDL that contain apoA-IV or apoE are significantly larger than (A-I) HDL or (A-I/A-II) HDL. It has been demonstrated that ApoA-IV has anti-oxidant, anti-inflammatory and anti-atherosclerotic actions in vivo [9,32].
The anti-atherosclerotic activity of apoE, related to receptor-mediated uptake of lipoprotein is well known [33]. Both murine and human apoE can inhibit the development of atherosclerosis in mice without any significant effect on hypercholesterolaemia. Moreover, apoE has been shown to have allele-specific anti-oxidant activity. ApoE stimulates eNO release and has anti-inflammatory activities [35,36].
ApoJ is associated with a subset of small HDL containing PON1 enzyme [37]. It has been reported that HDL-associated apoJ can inhibit LDL oxidation by artery wall cells. ApoJ is cytoprotective at low physiological levels [38].
Minor proteins associated with HDL reaches as many as 75, as demonstrated in recent proteomic studies [39]. In addition to proteins, HDL carries a number of small peptides of between 1 to 5 kDa in mass [40]. Indeed, more than 100 peptide components can be identified in HDL [41]. These peptides are present in HDL at low concentrations of about 1% of total HDL protein, with some representing fragments of larger proteins. The associations of small peptides with HDL, as a vehicle may represent a pathway for peptide delivery or scavenging in order to slow renal clearance and proteolysis, and a significant reservoir of plasma peptides for diagnostic evaluation [42].
The remaining protein mass is made up of minor amphipathic proteins, such as , apoD and apoM, enzymes such as PON 1 and PAF-AH, GPx1, and lipid transfer proteins such as LCAT and CETP and other proteins and peptides, such as SAA(a major positive acute-phase reactant), component of complemant, α1-antitrypsin(a potent inhibitor of serine proteinases), or amyloid [9,40].
PLTP plays an important role in the regulation of HDL metabolism [43]. Most tissues show expression of PLTP mRNA but liver and adipose tissue are probably the major contributors to plasma PLTP. PLTP is rather a non-specific lipid transfer protein. Several studies have shown that it is able to transfer all common phospholipid types [43,44]. The regulatory role of PLTP is achieved via its two main functions, phospholipid transfer activity and the ability to modulate HDL size and composition in a process called HDL remodeling [45,46]. These include the cellular receptors SR-B1 and ABCA-1 as well as plasma proteins such as CTEP, LCAT and endothelial-bound enzymes LP and HL [47]. PLTP is able to generate prebeta-HDL particle through HDL remodeling and has also a major role in maintaining plasma HDL levels owing to its ability to transport surface remnants produced by lipolysis of TG rich lipoproteins. Several study results suggest that PLTP may act as pro-atherogenic, rather than an anti-atherogenic factor in vivo [45-49]. Elevation of PLTP activity results in HDL hyper catabolism and enhanced hepatic VLDL-secretion [49,45].
CETP is encoded on chromosome 16 and primarily expressed by the liver and adipose tissue [50]. In the circulation, CETP facilitates the bidirectional transfer of cholesteryl esters and TG between HDL and (V)LDL. CETP may also undergo conformational changes to accommodate lipoprotein particles of different size, such as HDL. On the other hand, enlarged HDL in patients with CETP deficiency or after treatment with CETP inhibitors had increased capacity for cholesterol efflux [50,51].
Phospholipids of the HDL are phosphatidylcholine, sphingomyelin, phoshatidylinositol, phospahatidycholine, lysophosphatidy ethanolamine ceramide, phosphatidylglycerol, phosphatidylserine, and cardiolipin.
2.1.4. Discoidal HDL and Spherical HDL
Recent studies have addressed the hypothesis that the multiple biological functions of HDL are mediated by distinct particle subspecies defined by specific cluster(s) of bound proteins.
Emerging that, HDL subpopulations are also functionally heterogeneous and that they vary in terms of their ability to protect against CVD (Table 2). As the cholesterol esterifies, it seeks the innermost part of the forming HDL particles which cause the particle to go from discoidal to spherical. As the HDL particle matures it is no longer called preβ, but rather α-1, 2 or 3 [52-54].
Factors Leading to HDL Heterogeneity
| CETP and PLTP | Generates lipid-free/lipid-poor ApoA-I and remodels spherical HDL into large and small particles. |
| LCAT | Discoidal HDL→spherical HDL, Generates lipid-free/lipid-poor ApoA-I |
| HL | Large TG rich-HDL→small spherical -HDL Generates lipid-free/lipid-poor ApoA-I. |
| EL | Small spherical -HDL→smaller HDL particles. Does not generate lipid-free/lipid-poor ApoA-I. |
| SR-B 1 | Spherical -HDL→smaller HDL particles. Does not generate lipid-free/lipid-poor ApoA-I. |
Discoidal HDL biogenesis involves the association of lipid–free or lipid-poor apolipoproteins with phospholipids and unesterified cholesterol that are exported from cell membranes via the ABCA1 [55,56].
The lipid-free or lipid–poor apolipoproteins that are involved in HDL biogenesis either originate from liver or intestine. They dissociate from triglyceride-rich lipoproteins that are undergoing lipolysis by LP or, in the case of apo A-I, the main HDL apolipoproteins dissociate from mature spherical HDL that are being remodeled by various plasma factors [57].
2.1.5. Subclasses of HDL
HDL particles can be separated by several laboratory techniques; ultracentrifugation, nuclear magnetic resonance spectroscopy, and electrophoresis, just to name a few [8,9]. Plasma HDL particles are highly heterogeneous in their physicochemical properties, intravascular metabolism and biologic activity [40].
The two major categories of HDL particles separated by electrophoresis are pre-β and alpha. After α-HDLs mature, they deliver the cholesterol to steroidogenic tissues (mostly adrenal) and adipocytes. Phospholipidated ApoA-I is sometimes referred to as prebeta-1 HDL. Disciodal HDL and lipid-free/lipid-poor apoA-I, migrate to a pre-β-position. Spherical HDL containing apoE as the only apolipoprotein migrate to a γ-position [40].
At present non-denaturing gradient gel electrophoresis is the most sensitive approach for quantifying the size distribution of HDL subpopulations. This technique has been used to classify HDL into 5 distinct subpopulations on the basis of average diameter [40,58,59]; HDL2b(10,4 nm),HDL2a(10,3 nm),HDL3a(9,9 nm), H-DL-3b(8,0 nm) and HDL3c(7,3 nm) (Fig. 1).
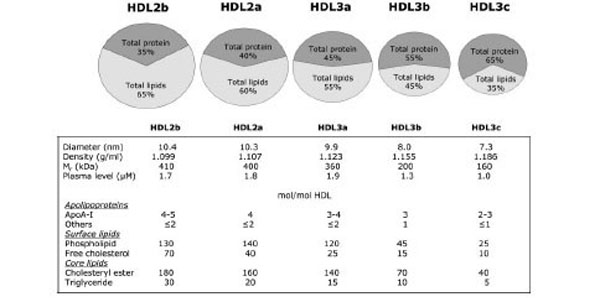
Plasma HDL particles are highly heterogeneous in their physicochemical properties, intravascular metabolism and biologic activity.
Of the all HDL subpopulations, the largest α-migrating HDL is the best negative predictor of recurrent cardiovascular events, while the smaller, α-migrating HDL is emerged as a positive, rather than a negative predictor [58].
2.1.5.1. HDL2, HDL3 and CVD
Proton NMR spectroscopy is used widely to quantify HDL subpopulation size and concentration in clinical settings [60]. The HDL in human plasma consists of two main sub-fractions HDL2 and HDL3. HDL2 are considerably larger and less dense than HDL3 [61]. When subjected to agarose gel electrophoresis, spherical (A-I) HDL and (A-I/A-II) HDL migrate to an α-position [62, 63]. ApoA-I is found more in HDL2, whereas apoA-I: A-II is found more in HDL3. For example, immune-isolated apoA-I: A-II is predominantly found in the HDL3, whereas apoA-I is a prominent component of both HDL2 and HDL3 [40].
The HDL2 subfraction contains mainly (A-I) HDL, whereas (A-I/A-II) HDL are largely, but not numerous insights into HDL functionality [64,65]. In addition to being a source for better markers of cholesterol flux, HDL has pleiotropic functions that include anti-inflammatory, antioxidant, antithrombotic, and nitric oxide–promoting effects [1].
Proteins that were preferentially associated with the dense HDL3 subpopulation included apoL-I, apoF, apoA-IV, apoM, apoJ, apoD, PON1, PON3, PLTP, A1AT, SAA, albumin, fibrinogen, haptoglobin-related protein(Hrp), platelet basic protein (PBP) and transthyretin [9,40].
Numerous population studies have suggested that HDL2 may be more cardioprotective than HDL3 [1]. However, there are also inconsistencies, with reports suggesting that HDL2 and HDL3 are equally cardioprotective [64,66]. For example, HDL3 have been reported to inhibit expression of VCAM-1 in cytokine-activated human umblical vein endothelial cells more effectively than HDL2. On the other hand, HDL2 and HDL3 appear to be equally effective at exporting cholesterol from cells that express ABCG1. Importantly, SAA selectively impairs cholesterol efflux properties of small, dense HDL3 particles. HDL2 and HDL3 are also indistinguishable in terms of their ability to reduce cholesteryl ester hydroperoxides to the corresponding, less deleterious, cholesteryl ester hydroxides [67,34].
Smaller and more dense HDL, especially HDL3b and HDL3c, have been reported to inhibit the oxidation of LDL more effectively than HDL2 [68-70].
In light of recent developments, there is a growing need to identify other HDL-related subclasses and functions, and to find biomarkers that will better predict cardiovascular risk and biomarkers that can be used to assess the clinical benefits of novel HDL-targeted therapies.
3. BIOLOGIC FUNCTIONS OF THE HDL
HDLs perform functions including several immunological activities (Table 3) [71]. The other atheroprotective functions of HDL that have more recently attracted attention among other actions include, its anti-apoptotic, anti-thrombotic and anti-infectious functions (Fig. 2) [72,73].
HDLs Perform Functions Including Several Immunological Activities
| anti-oxidative | inhibition of LDL oxidation | anti-inflammatory |
| anti-thrombotic | inhibition of VCAM-I expression | cholesterol- PL efflux |
| anti-apoptotic | inhibition of endothelial cell apoptosis | vasodilatory |
| anti-infectious | inhibition of platelet aggregation | LPS-binding |
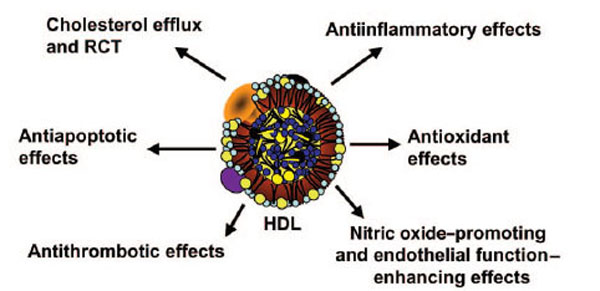
Several antiatherogenic properties of HDL.
HDL classically functions in RCT, removing cholesterol from peripheral tissues and delivering it to the liver and to steroidogenic organs, by the binding of the major HDL apolipoprotein apolipoprotein A-I (apoA-I) to the high affinity HDL receptor (SR-BI) [74]. Nascent HDL can promote cholesterol efflux from macrophages via ABCA1 and mature HDL via ABCG1. Polymorphisms of ABCA1 greatly affect HDL-C and are major determinants of a person’s HDL-C level [75]. Cholesterol is esterfied to CE in the plasma by LCAT, and both unesterified FC and CE are transported to the liver either directly via SR-BI or indirectly via transfer to apoB-lipoproteins by the action of CETP [76]. The enzyme LCAT esterifies the cholesterol causing formation of larger, mature α-HDLs after delipidation (by SR-B1) [1,77]. The now smaller α or pre-beta HDLs return to tissue ABCA1 for relipidation. HDL content is modulated by HL, EL, and PLTP [78].
3.1. 2+2=4? Not Always….: the Torcetrapib Disaster
The torcetrapib experience is the quint essential example of our inevitable failure, when the fair but complicated mechanisms of the human body are underestimated. Researchers should have been very excited to develop drugs to increase HDL. Torcetrapib, an inhibitor of the CETP, was a pioneer. Torcetrapib had been shown to substantially increase HDL concentrations by 50%-100%. Unfortunately, the party turned to be a nightmare [79-82]. The large clinical outcome trial investigation of “Lipid Level Management to Understand its Impact in Atherosclerotic Events” was terminated early because of increased cardiovascular morbidity and total mortality despite an observed 72% increase in HDL concentrations in individuals treated with the drug [82]. Nevertheless, the Torcetrapib experience urged us to ask ourselves the crucial questions with answers that finally lead us to the contemporary concept of “HDL functionality”. Afterall, it is clear that therapeutic optiones based solely on increasing HDL may not be adequate for cardiovascular risk reduction. So, HDL-related atheroprotection remains complex and not fully understood. The search is continuing to find the best approaches for the prevention of the loss or the restoration of the anti-oxidant and anti-inflammatory potential of HDL [83,84].
3.2. HDL: Trucks for the “Good” Cholesterol
The cholesterol efflux capacity of HDL particles is related to their ability to remove cholesterol from membranes of peripheral cells and particularly macrophages and foam cells via interaction with the ABCA1 and ABCG1 transporters and/or SR-BI receptor [85, 86].
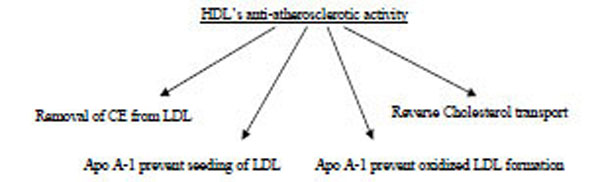
HDL thus performs this clean-up by acting as a garbage truck, picking up its garbage in the arterial wall and transporting it to the liver.
It is well known that, atherosclerotic lesion development is initiated by the accumulation of cholesterol from LDL in macrophages of the arterial wall. HDL exerts its protective function by removing excess cholesterol from macrophages in the arterial wall and transporting it to the liver, where it can be excreted from the body [40]. In contrast to lipid-free apolipoproteins, lipid-containing HDL particles induce both specific and nonspecific forms of cholesterol efflux. The PL content of HDL is an important determinant of such SR-BI-mediated cholesterol efflux [40].
Another mechanism implicated in HDL-mediated cholesterol efflux is retroendocytosis [87]. Finally, HDL mediated cholesterol efflux from macrophages may be facilitated by apoE secretion. Indeed, macrophage-derived apoE can associate with HDL and improve its cholesterol acceptor properties. ApoE possesses established anti-atherosclerotic activity, which is normally ascribed to its lipid transport properties. In fact, apoE possesses distinct anti-oxidative properties and can promote regression of atherosclerosis independent of lowering plasma cholesterol levels [40, 88, 89].
It has been shown that PON1 enhances cholesterol efflux from macrophages by promoting HDL binding mediated by ABCA1 [90]. The cholesterol efflux capacity of HDL is considerably impaired during inflammation [86]. In vitro studies have demonstrated that MPO-catalyzed oxidative modification of HDL or apoA-I leads to the loss of ABCA1-dependent cholesterol efflux function of the lipoprotein and converts HDL into a pro-inflammatory particle [91]. Enrichment of HDL with SAA results in increased HDL binding to macrophages, decreased cholesterol efflux from macrophages, and increased selective uptake of CE by macrophages [93].
Extensive research focus on strategies to increase the transport of cholesterol via HDL to the liver. Very likely, these studies will be the background of the future medications.
3.3. Importance of the HDL Receptor for the Protective Function of HDL
A specific receptor that mediates the uptake of cholesterol from HDL by the liver is essentially required for the atheroprotective function of HDL. Researchers of the laboratories have focused for more than 10 years on the function of the receptor for HDL, SR-BI. In mice this receptor is essential for the delivery of HDL cholesterol to the liver. Mice that lack this receptor accumulate large amounts of HDL cholesterol in their blood, but still rapidly develop extensive atherosclerosis as HDL cannot function properly. The mutation in SR-BI leads to a high HDL cholesterol level in the blood and proves that SR-BI is also essential for the uptake of HDL cholesterol in humans [93,94].
3.4. Antithrombotic Effects of HDL
Platelet aggregation is inversely correlated with HDL levels in humans, suggesting that HDL has antiplatelet actions. HDLs display an antithrombotic effect by inhibiting platelets aggregation, reducing von Willebrand factor levels, and enhancing the activity of activated protein C and S [95-98]. Mechanistically, HDL may act indirectly on platelet activation via effects on endothelial cell, although may expose a direct effect [99].
The administration of reconstituted HDL to humans or the infusion of apoA-I Milano into rats inhibits platelet aggregation, further supporting the concept that HDL inhibits platelet activation in vivo [98]. In a rat model of acute arterial thrombosis, the infusion of apoA-I Milano caused a prolongation in the time of thrombus formation and a reduction in the weight of the thrombus suggesting that HDL inhibits thrombus formation in vivo within a time frame relevant to acute thrombosis [100].
Prostacyclin acts synergistically with NO to induce VSM relaxation, inhibit platelet activation, and diminish the release of growth factors that stimulate the local proliferation of VSM cells [98]. In addition to promoting NO production, HDL causes enhanced prostacyclin synthesis, which can modify thrombosis as well as other intravascular events [98].
There is a positive correlation between plasma apoA-I levels and anticoagulant response to APC/protein S in vitro [100]. There is also an inverse correlation between HDL and plasma thrombin activation markers in vivo, and an inverse correlation between HDL and plasma thrombin activation markers such as prothrombin fragments in vivo; suggesting that HDL may modify thrombin generation under normal conditions. HDL, particularly HDL2 or large HDL [101], functions as an anticoagulant cofactor when it enhances the inactivation of purified coagulation factor Va by APC and protein S in vitro, which inhibits thrombin generation and fibrin clot formation [98].
Because thrombomodulin is both an antithrombotic factor via the generation of APC and an antiinflammatory factor via currently unknown mechanisms, the upregulation of thrombomodulin expression by HDL may have a variety of important implications regarding HDL action in humans [102].
HDL Sphingolipids and Antithrombotic Activities:
HDL transports various sphingolipids that are present in plasma in the micromolar range, and at least 4 kinds of HDL sphingolipids may directly or indirectly contribute antithrombotic activity [102].
Glucosylceramide → ↑cofactors for APCsphingosine → ↓procoagulant interactions between factors Xa and Valysosphingolipids → ↓vasoactive and antiapoptotic activities (reducing endothelial cell apoptosis)
Lysosphingolipids → ↓endothelial cell adhesive reactions
HDL and Fibrinolysis:
HDL→↓ PAI-I→ fibrinolysis↑
The oxidation of HDL alters the influence of HDL on fibrinolysis because oxidized HDL3, but not native HDL3, promotes PAI-I expression and consequently suppresses fibrinolysis [98].
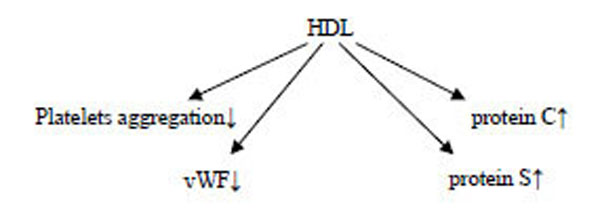
Futher, HDLs seem to exert a beneficial effect on endothelium function by modulating the activity and/or production of a number of molecules and affecting vascular tone and thrombogenicity [102].
3.5. Anti-oxidant Functions of HDL
Normal functional HDL has high levels of anti-oxidants and active anti-oxidant proteins and enzymes with high anti-oxidant potential and has anti-inflammatory activity [92,97]. Many pathological processes associated with systemic inflammation are characterized by the presensce of dysfunctional or pro-inflammatory HDL, including CVD, migraine, metabolic syndrome, chronic kidney disease, infections and some rheumatological diseases [103]. In most species there is a reduction in HDL and apoA-I levels with a concomitant loss of anti-inflammatory potential [10].
Indeed, anti-oxidative deficiency of HDL relative to LDL oxidation by artery wall cells is observed in the acute phase, concomitant with decreases in the activity of PON1 and PAF-AH [92].
HDL can directly inhibit oxidation of LDL; or other targets containing phospholipids. In addition, inhibition of oxidative events and oxidative stress in vivo may be achieved indirectly via other functions of HDL, such as induction of cholesterol efflux and, in general, via ‘anti-inflammatory’ functions of HDL [34].
It has been shown that circulating HDL accumulates oxidized phospholipids, such as hydroperoxides, lyso-PC and F2-isoprostanes [34,104].
Recent evidence further links the interaction of HDL, particularly apoA-I with ABCA1 and ABCG1 as components of a pathway that could suppress inflammation [105]. Enhanced remodeling, likely results in the liberation of lipid-poor apoA-I that could enhance cholesterol efflux and therefore be athero-protective. At the same time, lipid-poor apoA-I is subject to rapid catabolism [7]. HDL cholesterol activates endothelial cell production of the atheroprotective signaling molecule NO, and it promotes endothelial repair [106].
Atherosclerotic vessels → ↑superoxide anion →↓eNOS →↓NO→↑↑neutrophil adherence to endothelium,smooth cell proliferation,platelet aggregation and adhesion.
This may be related to decreased HDL-associated PON1 activity [107]. OxLDL causes depletion of caveolae cholesterol in endothelial cells via the scavenger receptor CD36, leading to eNOS redistribution to an intracellular locale and an attenuated capacity to activate the enzyme [98]. This process may play a critical role in the early stages of hypoactivity of PON enzyme-induced vascular disease, and interestingly, later in the pathogenesis of atherosclerosis, when oxidized phospholipids have accumulated in the arterial wall [106].
Furthermore, the OxLDL-induced fall in caveolae sterol content is prevented by cotreatment with HDL [106]. Thus, in the presence of OxLDL, one of the actions of HDL that occurs through SR-BI is to preserve the unique lipid environment within caveolae, thereby maintaining the normal subcellular localization and function of eNOS [106,108,109].
The transfer of oxidized phospholipids to HDL may be followed by subsequent degradation of oxidation products by HDL enzymes or by delivery to the liver for degradation [34]. Hydrolysis of oxidized phospholipids in HDL leads to the destruction of pathological activity [34]. The antioxidative activities of HDL might equally become impaired in the presence of inflammation due to the replacement of Apo A-I by SAA and altered enzymatic activities [92,110]. All these mechanisms might limit the capacity of HDL to inactivate oxidized phospholipids, resulting in their elevated accumulation in LDL [92,104]. The interaction of apoA-I with cholesterol loaded macrophages suppress LPS-induced production of inflammatory cytokines [111].
Oxidation and modification of HDL are through; non-enzymatic glycation, homocysteinylation, reactions with metal ions, peroxyl radicals, hydroxyl radicals, aldehydes, MPO-generated oxidants, elastase, lipoxygenase, phosphor-lipase A2.
HDL from diabetic subjects has evidence of glycated apoAI and apoAII, and this glycated apoAI has altered structure and lipid binding activity [12]. Similarly, HDL incubated with high glucose results in a reduction in PON1 activity and decreased anti-inflammatory activity [11]. A prominent factor that can lead to HDL dysfunction is the post-translational modification of apoAI [12]. In addition, Malondialdehyde modification of HDL leads to similar changes in regard to HDL structure and function, and is associated with the loss of lysine and tryptophan residues, and apoAI polymerization [12,112]. Copper mediated oxidation of HDL leads to altered HDL migration on an agarose gel, apoAI proteolysis, and decreased ability of HDL to unload cholesterol esters from cholesterol loaded macrophages [12]. Lipid peroxides added to human plasma can covalently modify apoAI [12]. Also, incubation of HDL with activated neutrophils was shown to render apoAI more negatively charged and decrease its cellular cholesterol acceptor activity [112].
The transfer of oxidized phospholipids to HDL serves three purposes. In the circulation a 74 kDa CTEP protein shuttles between HDL and apoB –containing lipoproteins and facilitates the bidirectional transfer of CE and TG between HDL and (V) LDL. CTEP may undergo conformational changes to accommodate lipoprotein particles of different size such as HDL, LDL and VLDL [40].
- The transfer of lipid hydroperoxide from ‘seeding molecules’LDL prevents the initiation of a free radical chain reaction of oxidation.
- Some of the more advanced products of phospholipid oxidation serve as ligands for SR-B1 and promote uptake of modified lipoproteins by macrophages as well as prothrombotic effects mediated by platelet scavenger receptor CD36.
Interestingly, when oxidized phospholipids are present in HDL (oxidized HDL), the receptors that recognize the particle may be different and the effects may be opposite to those of oxidized LDL. - Some of the products of oxidation are chemically reactive and can modify lysine residues in apoB [40].
3.5.1. Paraoxonases 1 and 3 of HDL
Multifunctional enzyme (PON1) is an HDL-associated esterase/ lactonase, which derived its name from one of its most commonly used in vitro substrates, paraoxon. PON1 exerts a protective effect against oxidative damage of cells and lipoproteins and modulates the susceptibility of HDL and LDL to atherogenic modifications such as homocysteinylation [113].
HDL has the ability to inactivate oxidized phospholipids, in part due to PON1 activity but potentially also due to other enzymes or properties of HDL. In general, it has been shown that multiple factors (genetic, pathological, physiological, pharmacological and lifestyle) can change the PON1activity (very similar to the HDL). HDL and less efficiently, VLDL but not LDL promote PON1 secretion from cells. Furthermore, a pro-atherosclerotic high-fat diet, smoking, sedentary life style and obesity lead to reduced PON1 activity(or reduced HDL–C). Dietary fatty acids act by the modulation of PON1 gene expression and activity. PLTP activity is increased in subjects with several risk factors for atherosclerosis .It increases with age, BMI, cigarette smoking and type 1 and type 2 diabetes mellitus [114,115].
However, it should be noted that the role of PON1 in the direct protection of LDL from oxidative stress is not firmly established at present [114]. The molecular mechanisms involved include an effect on PON1 hepatic synthesis or secretion and/or modification of PON1 interactions with HDL. Such as, PON1 interaction with apoA-I is critical for enzyme stability. Moreover, signs of oxidative stress, vascular inflammation and thrombotic tendencies have been observed in PON1-deficient mice [116].
Interestingly, PON1 has been reported to catalyze the hydrolysis of a variety of lactones, including homocysteine thiolactone, suggesting that its native activity is as a lactonase [114]. Such a PAF-AH/PON1-coupled protective function of apoA-I can effectively divert proatherogenic LOOH to less harmful products [12].
Chronic pathological conditions associated with oxidative stress, such as chronic renal failure, rheumatoid arthritis, migraine and Alzheimer’s disease, are frequently associated with reduced activity of PON1 [103,113,114]. In human studies, higher PON1 activity is associated with a lower incidence of major cardiovascular events [114]. However, it remains uncertain whether this relationship is causal or correlational.
Paraoxonase 3 is also associated with HDL and has been shown to prevent the oxidation of LDL in vitro [114,117]. Furthermore, human PON3 transgenic mice have been shown to be protected from the development of atherosclerosis, without any significant changes in plasma lipoprotein cholesterol, triglyceride or glucose levels [34].
Thus, PON1 activity is a promising biomarker of HDL function and cardiovascular risk independent of HDL concentrations [1].
3.5.2. Platelet-activating Factor Acetylhydrolase
Equally termed LPPLA2, PAF-AH is a calcium independent hydrolytic enzyme that degrades platelet activating factor by hydrolyzing the sn-2 ester bond to yield biologically inactivate lyso-PAF [40] .PAF-AH is another enzyme that is found in HDL that can hydrolyze oxidized phospholipids [118,119]. Lipoproteins isolated from mice expressing human PAF-AH are more resistant to oxidative stress [34]. Moreover, murine HDL with human PAF-AH has been shown to inhibit foam cell formation and facilitate cholesterol efflux in macrophages [34].
In humans, PAF-AH deficiency is associated with increases in CVD [120]. Whether PAF-AH plays a causal role in CVD or simply is increased in response to the chronic inflammatory environment needs to be established. It is believed that PAF-AH in the necrotic core of coronary lesions may contribute to inflammation and plaque vulnerability. For example, prolonged inhibition of PAF-AH with the oral inhibitor Darapladib prevented necrotic core expansion in patients with CVD [34,120].
There are data suggesting that PAF-AH, rather than PON1, is the major hydrolase in HDL responsible for the hydrolysis of oxidized phospholipids [121].
3.5.3. Glutathione Peroxidase 1
GPx-1, another enzyme detected in HDL, can reduce lipid hydroperoxides to corresponding hydroxides and thereby detoxify them. Multiple clinical studies suggest an atheroprotective role for GPx-1. It seems that the role of GPx-1 in the development of atherosclerosis is particularly prominent under conditions of significant oxidative stress [34].
3.5.4. Lecithin: Cholesterol Acyltransferase
Lecithin:cholesterol acyltransferase has been shown to directly hydrolyse oxidized polar phospholipids [122,123].
3.5.5. Sphingosine-1-phosphate
HDL is the most prominent plasma carrier of S1P [124]. Moreover, many biological effects of HDL, such as the induction of endothelial NO production, vasodilation, survival and cardioprotection, are partially mediated by S1P [125]. In general, multiple effects of HDL on endothelial cells, such as migration, proliferation, endothelial integrity and angiogenesis, are mediated in part by S1P [73]. Furthermore, S1P in HDL inhibits pro-inflammatory responses, such as the generation of ROS, activation of NAD(P)H oxidase and the production of monocyte chemoattractant protein-1, in vascular smooth muscle cells and the aorta [124].
In endothelial cells, S1P in HDL inhibits tumour necrosis factor-α-induced expression of cell adhesion molecules and promotes prostacyclin production [125].
Another important anti-inflammatory role of HDL is to limit the expression of cytokines such as tumor necrosis factor-alpha, and interleukin-1 that mediate up-regulation of leukocyte endothelial adhesion molecules [126].
4. RECENT CONCEPTS
4.1. What if HDL Fails in its Mission? The New Definition:”Dysfunctional HDL”
The concept of HDL dysfunction was originally developed by scientists in the mid-1990’s [12]. Researchers have proposed that in some people HDL was dysfunctional, or malformed, and unable to do its job properly. HDL particles have several functions related to trafficking cholesterol and proteins. If HDL particles perform these biologic tasks they are termed, “functional”. If they do not, they are termed “dysfunctional” [12].
Few studies are available in human populations investigating involvement of vascular inflammation and oxidative stress-related dysfunctional transformation of HDL in establishing CVD. Nearly half of people who get heart attacks have healthy cholesterol levels. Apparently, not all HDL, often referred to as "good cholesterol", protects against plaque build-up in artery walls [40].
The potent atheroprotective properties of HDL particles originate from their unique composition and structure. As a result, HDL may lose its normal function and acquire pathological functions. For one, dysfunctional HDL is not that common, plus population studies show that people with high HDL usually have lower risk for heart disease. Whereas some people who have high HDL levels still get heart attacks and suffer from other CVD [127].
There are many possible alterations between dysfunctional HDL and normal functional HDL. One possibility is a change in the protein composition of HDL. Human HDL particles are quite heterogeneous, encompasssing a range of sizes and densities [40].
The close association between inflammation, oxidative stress, dyslipidemia, and atherosclerosis suggests that such HDL alterations play a significant role in disease progression. As a result, HDL particles progressively lose normal biological activities and acquire altered properties [127].
However, when anti-oxidant and anti-inflammatory functions of HDL are overwhelmed by pathological processes, such as inflammation, HDL is converted into a ‘dysfunctional’ pro-inflammatory particle. With respect to atherosclerosis, if an HDL does not perform anti-atherogenic functions, it is called a “pro-atherogenic” HDL [92].
HDL is known to undergo dramatic modification in structure and composition as a result of the concerted actions of the acute-phase response and inflammation. This ‘dysfunctional’ (HDL-D) is characterized by decreased levels and activities of anti-inflammatory and anti-oxidant factors, such as apoA-I and PON1 [38].
The dysfunctional HDL contains oxidized phospholipids and lysophospholipids, as well as pro-inflammatory proteins, such as SAA and ceruloplasmin. Eventually, functionally it cannot promote cholesterol efflux effectively or prevent LDL oxidation [34].
4.2. The Conversion of HDL to Proinflammatory HDL. Way to the Dark Side
Alterations occurring in HDL composition and metabolism due to inflammation are intimately associated with impaired biological activities. According to recent studies in patients with CVD, HDL is not only ineffective as an anti-inflammatory and antioxidant but is actually a pro-inflammatory and pro-oxidant promoting LDL oxidation (Fig. 3) [92].
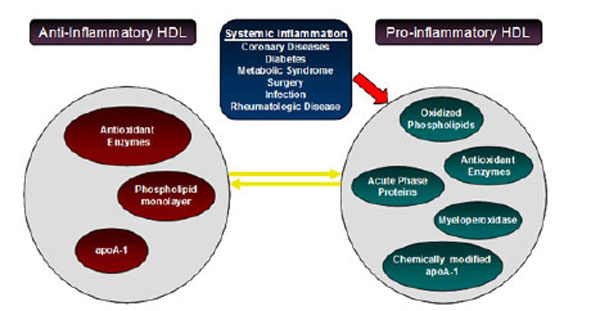
Alterations occurring in HDL composition and metabolism. Inflammation converts HDL to a particle that is not only ineffective as an anti-inflammatory and antioxidant but a pro-inflammatory and pro-oxidant, promoting LDL oxidation.
In acute and chronic inflammation (e.g .influenza A), the content and functions of HDL can change drastically converting atheroprotective HDL to pro-atherogenic HDL [128]. During the acute phase response to infection, inflammation or trauma, cytokine mediated changes in the composition of plasma proteins occur. HDL levels are dramatically reduced and HDL composition is altered by converter agent (e.g, endotoxin, cytokines, MPO, CRP, SAA). SAA proteins are major acute–phase reactants secreted during the acute phase of the inflammatory response [91,128].
The synthesis of SAA and group IIA secretory phospholipase A2 in the liver is stimulated by circulating cytokines (IL-1β, IL-6, TNF-α) during the acute phase response. Endotoxin treatment has been associated with an increase in SAA expression and a reduction in efflux from macrophages to acute phase-HDL [7].
Inflammation induces major changes in HDL levels and composition. HDL from many CVD patients was found to be pro-inflammatory, thus increasing monocyte chemotaxis in response to LDL, unlike the HDL from healthy controls that reduced monocyte chemotaxis [12].
An oxidative environment (e.g.subclinical inflamation) is produced when an acute phase response (TNF-α and IL-6) occurs as a result of nonspecific immunity. Thus, HDL appears to be part of the innate immune system and can be either proinflammatory or anti-inflammatory depending on the presence or absence of an acute-phase response and systemic inflammation. As part of the acute-phase response, activities of HDL-associated enzymes including PON1, PAF-AH, LCAT, CETP, and PLTP can be compromised, made dysfunctional or both [92,129].
MPO is a key inflammatory mediator of macrophages and other leukocytes, and systemic inflammation is thought to convert HDL to a dysfunctional form that loses its antiatherogenic effects [91]. MPO is enriched in human atheroma and its presence may promote lesion progression, by increasing LDL oxidation, and block plaque regression, by modifications of apoAI/HDL that impair RCT [12]. It has been recently demonstrated that incubation of HDL with MPO, in a chlorinating reaction (MPO/H2O2/Cl−) that produces HOCl , or with reagent HOCl, leads to a loss of unsaturated fatty acids in phospholipids and cholesterol esters, and the loss of cholesterol acceptor activity. In addition to decreased lipid acceptor activity, another study also showed that MPO or HOCl modified HDL was more susceptible to uptake and degradation by macrophages, thus turning HDL from a lipid accepting lipoprotein to a lipid loading lipoprotein [12,91]. Thus, the site directed substitution and cholesterol efflux data indicate that neither methionine nor tyrosine serve as the oxidant sensitive residue, involved in MPO-induced apoAI inactivation, specifically the loss of alpha helix content, and the loss of tryptophan and lysine residues [12,91].
4.2.1. Conversion of Anti-inflammatory HDL Into Pro-inflammatory HDL by MPO
Products of an inflammatory enzyme MPO → MPO target main Apo A1 → Converting HDL into pro-inflammatory non-functional Apo A1 → macrophages retain increase cholesterol & cholesterol reverse transport reduced → MPO modify tyrosine amino acid moiety → dysfunctional HDL has increased hydroperoxidase → this makes it pro- inflammatory [12,91].
SAA is a pro-oxidant acute-phase reactant associated not only with disabling the anti-inflammatory role of HDL but also with creation of proinflammatory HDL [92] .It was shown that SAA containing acute phase-HDL is cleared faster and could contribute to the reduction in HDL levels. However SAA also has ABCA1 and SRBI-dependent cholesterol efflux capacity that could promote the maintenance of plasma HDL [92].
The hydrolysis of HDL by EL and sPLA2-IIA may be partly responsible for the reduction in HDL levels during the acute phase response [16].
The activity of PLTP, which transfers phospholipids between different lipoproteins, is positively associated with SAA and C-reactive protein in patients with low HDL and CVD. In contrast, CETP levels and activity appear to be reduced during inflammation in vivo [130].
Haptoglobin, binds to the core of HDL and that a defective Haptoglobin variant (Hp2-2), found in 40% of the population, may induce dysfunctionality in HDL [131].
α-1-antitrypsin is a potent inhibitor of serine proteinases. Its primary target elastase, but it also has moderate affinity for plasmin and thrombin. In the circulation ,α-1-antitrypsin is present exclusively in HDL as a 52 kDa protein. α-2-antiplasmin a serine proteinase inhibitor that inhibits plasmin and trypsin as two major targets and also inactivates chmotrypsin, in part, associated with HDL [132]. HDL carries inter- α-triypsin inhibitor heavy chain H4 and bikunin. Other proteolysis proteins detected on HDL include Hrp, kininogen-1, prothrombin, angiotensinogen and procollagen C-proteinase enhancer-2 [40].
In addition, HDL-associated proteins involved in complement regulation are represented by complement component C3, C4, C9 and vitronectin [40].
Although it was proposed as a compensatory mechanism (gene up-regulation) to prevent further reductions in HDL during this state, the studies report that exposure to vitamin E can restore HDL functionality and the process of reverse cholesterol transport [133].
4.3. Dsyfunctional HDL and HDL Associated Lipids
Another factor that might make HDL “dysfunctional” is a change in the HDL associated lipids [12]. PL constitute the major lipid class of HDL, build the surface lipid monolayer and ensure specific particle structure [12,40]. Typically, total plasma HDL contains 20-30 %( wt/wt) PL, 3-5% free cholesterol ,14-18% CE and 3-6 % TG[40]. HDL lipid composition might equally be altered during inflammation [92]. Enrichment in TG with depletion of CE in the HDL core is the most frequent abnormality of HDL lipid composition and occurs in hypertriglyceridemic states associated with decreased activity of lipoprotein lipase, hepatic lipase, LCAT, or a combination of these [89,92]. In addition, HDL triglyceride content can also be increased in hypertriglyceridemia as a consequence of elevated CETP activity [92]. CETP-mediated replacement of cholesteryl esters by triglycerides in the HDL core results in decreased plasma HDL-C levels, which is another feature of the acute-phase response [11,92].
Lipid peroxides may interfere with HDL’s antioxidant, anti-inflammatory, and cholesterol acceptor activities. HDL carries the bulk of lipid hydroperoxides in plasma [67]. In contrast, dysfunctional HDL might promote the transfer of lipid hydroperoxides to apoB containing lipoproteins and promote VLDL and LDL oxidation [12]. Minor bioactive HDL lipids include diacyglycerides, monoacylglycerides, free fatty acids, glycosphingolipipds, gangliosides, sulfatides and lysosphingolipids [40]. Among lysosphingolipids sphingosine 1-phosphate (S1P) is particularly intresting this bioactive lipid plays a key role in vascular biology and can function as a ligand for the family of G protein –coupled S1P receptors on endothelial and smooth muscle cells, which regulate cell proliferation, motility, apopotosis, angiogenesis, wound healing, and immune response. HDL is the major carrier of S1P in the circulation, ensuring bioavailability [40,134].
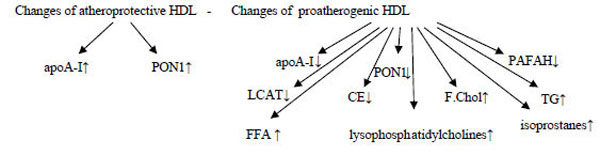
Insulin resistance→↑TG→CE-Poor HDL→very small HDL→ excreted by the kidneys → HDL↓
Inflammation and infection→ overexpression of endothelial lipase→HDL↓
Hepatic lipase deficiency→ larger HDLs↑
CETP deficiency→ very large HDLs↑ (CE remains in the HDLs).
5. LABORATORY MEASUREMENT OF HDL FUNCTIONALITY
What determines HDL functionality? There is no definitive answer to this question. It appears that there is no definitive common structural feature that determines HDL functionality. Complexity and costs of measuring all functions preclude these tests from becoming routine clinical laboratory tests for assessing the CVD risk for individual patient Table 4. Determining HDL function may also identify patients with normal or low HDL levels that are at particularly high risk for cardiovascular events. Generally, small HDL is more active in promoting cholesterol efflux and has greater anti-inflammatory and anti-oxidant properties [63]. However, increased small HDL in serum may indicate an aberration in the maturation of HDL and decreased RCT and maybe associated with increased risk of CVD [40,135]. The goal of identifying a population with dysfunctional HDL is the very possible benefit of these people from treatments such as apoA-1mimetics. Recognition of proinflammatory HDL might also justify traditional LDL-lowering approaches as statin therapy in this group, has given evidence that statin therapy also moderates HDL dysfunction [136-138]. Determination of HDL anti-inflammatory/proinflammatory function will likely yield important additional information beyond that available from simply knowing the quantitative level of HDL-C in an individual.
Available Laboratory Test to Measure HDL Function
|
Assays of HDL function |
Cholesterol Efflux and Reverse Cholesterol transport |
Cellular cholesterol efflux ex vivo.
Macrophage-specific RCT in vivo Centripetal cholesterol flux estimated from peripheral synthesis and uptake Mass fecal steroid excretion Kinetic modeling of cholesterol efflux from tissues in vivo by isotope dilution. |
|
Indexes of anti-Inflammatory activity |
Endothelial adhesion molecule expression Monocyte chemotaxis |
|
|
Indexes of antioxidant activity |
HDL-associated enzymes Paraoxonase Glutathione Selenoperoxidase Oxidized LDL and PL |
|
| NO Production and Endothelial Function | ||
| Metrics of Antiplatelet and Antithrombotic Activity |
Contrary to the regular assertions, serum HDL-C measurements might fall short in distinguishing risk groups in CVD. Evaluating HDL-C levels in quantity instead of quality may be deceptive and may be a factor in conflicted results in the literature [139,140]. Patients with a healthy lipid profile but experiencing a cardiovascular event pose a dilemma. A laboratory assessment heavily dependent on quantities can be extremely confusing particularly when dealing with a very heterogeneous structure like HDL. A great deal of data accumulates in literature that HDL loses its function as a post-traumatic modification caused by a plenty of impacts. Recent research point to the oxidative damage as the most probable culprit [12,40,92]. One can presume that measuring the body oxidative status is as valuable as measuring the dysfunctionality in HDL. The oxidative balance is a result of extremely complicated interactions, and trying to detect role players one by one is an unrealistic endeavor [38]. Commercially available, ready to use automatic laboratory instruments kits, measuring the body total oxidant and antioxidant status promise an exuberant welcome [141-144]. Although an indirect measure of HDL dysfunctionality, these tests will hopefully alleviate the stress on the issue. Oxidative status detection is intended to identify those in whom early interventions might reduce or ameliorate the disease. A routine panel of lipid parameters are about to be out of favor in this era of cost containment.
6. CONCLUSIONS
There are and will be considerable efforts to curtail CVD incidence. The ultimate goal is to identify markers of presumptive signs of the disease. Measurement of serum HDL in quantity was touted as a very sensitive test in general practice. Recent data urge the interpretation of HDL in the context of “functionality”. As we understand the structural components of HDL, and exact roles of each component in HDL function, traditional lipid panel studies will naturally be superseded by assays with greater sensitivity and specificity. Oxidative stress markers like PON1 or assays measuring total oxidant and antioxidant status are the most promising. Long term controlled studies focusing in determination of HDL quality are required and must be sustained.
CONFLICT OF INTEREST
The author(s) confirm that this article content has no conflicts of interest.
ACKNOWLEDGEMENT
None declared.
REFERENCES
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link] [PMC Link]
[PubMed Link] [PMC Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link] [PMC Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]

