All published articles of this journal are available on ScienceDirect.

Model of Abnormal Chromophore-Protein Interaction for Е181К Rhodopsin Mutation: Computer Molecular Dynamics Study
Authors Info & Affiliations
Abstract
The interaction of the 11-cis-retinal chromophore with the surrounding amino acid residues in the chromophore center of the rhodopsin protein has been investigated for the Е181К mutant form using molecular dynamics simulation. A comparative analysis of the arrangement of the amino acid residues in the chromophore center has been performed for both wild (native) and mutant rhodopsins. It is shown that for the Е181К mutant rhodopsin there is no proper binding of 11-cis-retinal with the surrounding amino acid residues. The distortion of the conformation states in the mutant rhodopsin molecule takes place in both the chromophore center and cytoplasmic domain. Our simulations suggest that a stable covalent linkage of 11-cis-retinal with the protein part (viz. opsin) of the rhodopsin molecule will not form. This, on the other hand, implies that the protein’s active site in the cytoplasmic domain, which is responsible for the G-protein binding (so-called transducin), may not be completely blocked.
Based on our molecular simulation data, we discuss the possible correlation between retinitis pigmentosa pathogenesis and the structural and functional properties of the rhodopsin protein.
INTRODUCTION
Rhodopsin is a typical member of the G-protein-coupled receptor family [1]. This visual pigment is the only light-sensitive protein in the rod outer segment photoreceptor membrane. It consists of the chromophore, 11-cis-retinal, and seven-helical membrane protein, opsin. 11-cis-retinal covalently binds to the protein through a protonated Schiff base (PSB) linkage to the ε-amino group of Lys296 [2]. In the dark-adapted state of rhodopsin, this PSB linkage is sufficiently stable. Light absorption initiates 11-cis-retinal isomerization, and then, rhodopsin photolysis. At the last stage of rhodopsin photoconversion, the transduction process is initiated. Finally, the disruption of the Schiff base linkage and removal of all-trans-retinal from the chromophore site of the protein moiety take place [3].
Amino acid substitution in the rhodopsin molecule during its biosynthesis leads to clinical pathology: the autosomal dominant form of retinitis pigmentosa (ADRP) [4–6]. Retinitis pigmentosa is an inherited degenerative disease of the retina characterized by progressive photoreceptor degeneration, night blindness, visual field constriction, and electroretinographic abnormalities, with rods affected earlier than cones [7]. Approximately 25% of ADRP is associated with the rhodopsin gene mutation RP4(RHO)/Rhodopsin(3q). More than 100 mutations of this visual pigment are known. These mutations are found in all the three rhodopsin domains (intradiscal, transmembrane, and cytoplasmic).
Amino acid substitution in the chromophore center of rhodopsin leads to the most clinically distinctive ADRP. These kinds of mutant forms belong to the II class of rhodopsin mutations, in which a stable PSB linkage cannot be generated and a stable molecule of the visual pigment cannot be formed [8, 9]. Mutations like these result in protein misfolding. Such visual pigment has a nonspecific activity [10, 11]. For example, the point mutations of Lys296, Glu113 or Glu181 lead to the permanent activity of the protein in vitro. In other words, rhodopsin is able to bind transducin in the absence of light and chromophore. This nonspecific activity in the dark could result not only in ADRP, but also other degenerative retinal diseases [12, 13].
Despite the fact that the genetic, biochemical, and biophysical studies of the rhodopsin mutant forms responsible for ADRP have been performed very intensely, the molecular mechanism of this pathology is not yet clear. Currently, there is no therapy that would stop RP evolution or restore the vision, so the visual prognosis is poor. Thus, understanding the molecular mechanisms of retinal degeneration caused by the above minimal structural changes in the photoreceptor has consequently become an aim of modern research.
In this work, we have used a non-traditional approach to investigate the initial stage of abnormalities in the visual processes caused by mutations in the rhodopsin molecule. The molecular dynamics simulation method has been applied to study protein misfolding as the consequence of mutations in the chromophore center of the rhodopsin molecule. We have analyzed the molecular dynamics of rhodopsin with the Е181К point mutation to understand the molecular mechanisms of the origins of retinal degeneration and retinitis pigmentosa.
MATERIALS AND METHODS
The dynamics of conformational changes in rhodopsin was calculated for three states: (1) rhodopsin containing no chromophore groups, i.e., free opsin; (2) wild rhodopsin with 11-cis-retinal; and (3) the rhodopsin mutant form E181K with 11-cis-retinal.
The initial configuration of the visual pigment was chosen by analogy with other reported computer simulation studies [14]. The rhodopsin molecule from the rhodopsin dimer model was used (PDB file 1HZX, chain A) [15]. To obtain free opsin, 11-cis-retinal was excluded from the initial rhodopsin molecule by computer manipulation; also, Glu181 was substituted by Lys181 in initial rhodopsin for obtaining the E181K mutant form.
Since the initial configuration of the molecule was incomplete (some parts of the PDB file were empty), preliminary calculations were carried out to determine the complete tertiary structure of the rhodopsin molecule before the simulations. Based on the primary rhodopsin structure [2], we found and minimized the missing fragments of amino-acid sequences 236–240 and 331–333, which had been embedded in the initial structure (PDB file 1HZX, chain A), using the MOE software package for computer simulations of biomolecules [16]. After that, the entire 1HZX molecule was minimized with missing fragments already embedded.
The next simulation stage involved a molecular-mechanical calculation of the molecule energy minimum and heating of the system from the crystalline state at T = 0 K to the physiological temperature of T = 300 K. The molecule structure that would correspond to the minimum energy at T = 300 K was searched for with very slow heating by about 20-degree steps in order to avoid strong system “oscillations” during the “heating”. After the system had been brought to T = 300 K, it took a few thousand “steps” (50–70 ps) for it to reach the equilibrium state with the minimum energy at this constant temperature. In fact, it is this equilibrium state with the minimum invariable energy of the system as a whole where the simulation process started. The moment of the simulation start was defined as the zero time.
The system temperature was kept constant (300 K) for three million steps using the Berendsen algorithm with a thermostat relaxation time of 0.2 ps [17]. An integration step of 1 fs was chosen for Newtonian motion equations; thus, the total simulation time amounted to 3000 ps.
The simulation of rhodopsin was carried out in an aqueous solution by means of AMBER 5.0 (Parm94) software package [18–20] and its modified version AMBER 7.0 (Parm96) for the MDGRAPE-2 special-purpose computer [21, 22].
The solvation of the system was done using the solvation procedure of the TIP3P water model in a specified spherical space [23].
The lengths of the bonds involving only hydrogen atoms were calculated using the standard SHAKE method [24].
All atomic interactions and trajectories were calculated and the entire rhodopsin molecule structure was determined. Simulations employed the Cornell atomic power field method [25]. The energy state of the system or the total interaction potential corresponded to the equilibrium state of the system, where the attraction forces were equilibrated by the repulsion forces. Different types of interaction that contribute to the stabilization of the biomacromolecule structure were taken into account:
U(r) =Σ Kr(r -req)2 is the potential of the intramolecular (valence) bonds;
+ Σ Kθ (θ-θeq)2 is the potential of the angular bonds (rotations);
+ Σ (1 + cos[nφ-γ]) is the potential of the dihedral (torsion) rotations;
+ Σ is the potential of the nonvalence (van der Waals) interactions;
+ Σ is the electrostatic potential.
The computation of the interatomic distances d with the coordinates of individual atoms i and j is presented in this work at specified time t: D1(x1,y1,z1)=D1(r1), D2(x2,y2,z2)=D2(r2), and, d = = |r2-r1|,
In other words, r1 and r2 are the spatial position vectors of atoms i and j, respectively, at a given time t (in the time range from t = 0 to t = 3000 ps).
RESULTS
We have performed molecular dynamics simulation of wild dark-adapted rhodopsin, free opsin, and the E181K mutant form. We have carried out a comparative analysis of amino acid residue arrangement in the PSB linkage area in all analyzed molecules.
Earlier we calculated the molecular dynamics of 11-cis-retinal and protein part of the visual pigment. It was found that the behavior of 11-cis-retinal correlates with both the displacements of the amino acid residues in the chromophore center and the structural deformations in the rhodopsin cytoplasmic domain, which is far enough from the chromophore center [26]. So we have performed the molecular dynamics simulation of amino acid residues not only in the chromophore center, but also in the rhodopsin cytoplasmic domain. It should be stressed out after the total molecular system (opsin+11-cis-retinal+water solvent) reach a relaxed state the 11-cis-retinal rearrangement has to occur at 0.4 ns. The time of 0.4 ns (viz., 400000 time steps) perhaps characterizes an adaptation time for the 11-cis-retinal inside the chromophore pocket and determines all subsequent events of rhodopin conformation. We have observed [26, 27] that the beta-ionone ring of the 11-cis-retinal begun to rotate with regard to the polyene chain axis to approximately 65º. This “twisting degree” we have calculated by comparing all the rotational torsion angles of all methyl groups C16-C20 of the 11-cis-retinal within the 3-ns dynamical changes. By other words, a “twisted” distorted chromophore configuration inside of rhodopsin binding pocket characterizes the differences between the original (crystal) and relaxed (liquid) of the rhodopsin protein thereby making a physiological sense [28]. It is also known that in the dark-adapted visual pigment, the chromophore, as an ligand antagonist, stabilizes the dynamics of α-helix H-VI, restricts its mobility, and prevents spontaneous activation of rhodopsin due to strong electrostatic interactions with Trp265 [3, 15, 29]. It follows from our model that not only Trp265 but also Tyr268 and Leu266 participate in the prevention of spontaneous activation of rhodopsin [26, 27].
The Interaction of 11-cis-retinal and Amino Acid Residues Glu113, Glu181, and Ser186 in the Region of the Protonated SCHIFF Base Linkage
The Wild Form of Rhodopsin
We have analyzed the molecular dynamics of the amino acid residues Glu113, Glu181, and Ser186, which take part in the stabilization of the PSB linkage in the rhodopsin molecule.
It is well known that Glu113 is the counter-ion of the PSB [30, 31]. The ionic bond (the salt bridge) between these groups stabilizes the interaction of α-helices H-III and H-VII. The disruption of this interaction can result in rhodopsin activation, i.e., rhodopsin becomes able to initiate transduction [32]. According to our calculations, the PSB linkage stabilization mechanism is rather complicated [33]. In these theoretical calculations, we have carried out a comparative analysis of the molecular dynamics of Glu113 in rhodopsin and in its apoprotein, free opsin. We have obtained unexpected results.
At the initial moment of modelling (t=0), the shortest distance between the N atom of the Lys296 ε-amino group and the O atoms of the Glu113 carboxy group in free opsin is ~3 Å (Fig. 1a). However, this distance increases to 5.5 Å after 1500 ps modelling. Up to the end of modelling (t=3 ns), the distances between different atoms of the Lys296 ε-amino group and Glu113 did not change. Although these amino acid residues are oppositely charged, they are not attracted to each other, as could be expected, but conversely, move away from each other.
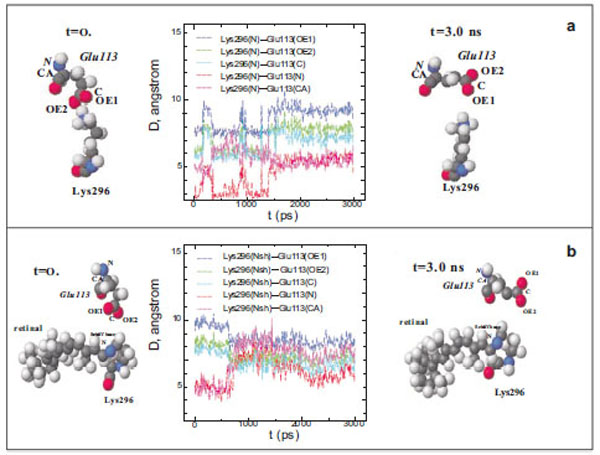
Diagram of interatomic distances (D) between different atoms of Glu113 and the ε-amino group N atom in Lys296, and the molecu-lar dynamics of Glu113 and Lys296 in free opsin (rhodopsin without the chromophore group) in the initial (t=0) and final (t=3ns) moments of modeling. The C, O, N, and H atoms are shown as grey, red, blue, and white spheres (a). Interatomic distances (D) between different at-oms of Glu113 and the protonated Schiff base N atom (NSh), and the molecular dynamics of Glu113 and Lys296 in rhodopsin at the initial (t=0) and final (t=3ns) moments of modeling (b).
The presence of the chromophore group, 11-cis-retinal, in rhodopsin changes substantially the molecular dynamics of Glu113 with respect to Lys296 (Fig. 1b). Initially, the distance between the O atoms of the Glu113 carboxy group and the N atom of the PSB is ~5 Å (t=0) rather than 3.3 Å, as in the rhodopsin crystal according to X-ray diffraction data [15]. After 600 ps modelling, this distance increases by 1.5 Å. The C and N atoms of the Glu113 amino acid residue that are involved in the formation of the peptide bond approach the N atom of the Schiff base by ~1.5 Å. This means that the negatively charged carboxy group of Glu113 moves away from the C=N bond of the Schiff base, while the α-helix H-III approaches this bond.
We have also analyzed the molecular dynamics of Glu181 and Ser186 amino acid residues because they are supposed to participate in the complicate molecular mechanism of PSB stabilization in rhodopsin. It was shown earlier that Glu181 becomes the counter-ion for the PSB after the formation of metarhodopsin I upon proton transfer from it to Glu113 through the system of hydrogen bonds involving Ser186 and water molecules [34].
The Glu181 position with respect to 11-cis-retinal remains almost unchanged on the time scale of the computer experiment. The distance between the carboxy group of Glu181 and the N atom of the PSB is ~6 Å (Fig. 2a; Fig. 3a), which coincides with X-ray diffraction data [15].
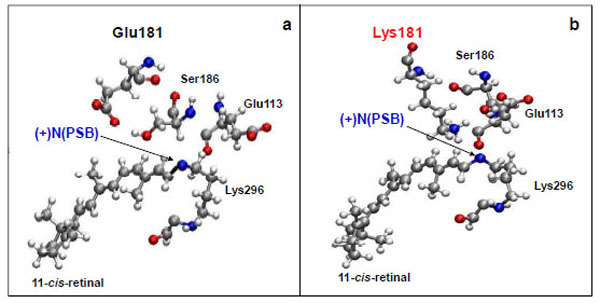
Molecular dynamics simulation of the amino acid residues Glu113, Glu181(Lys181), and Ser186 near protonated Schiff base link-age ((+)NPSB) in wild rhodopsin (a) and in the E181K mutant form of rhodopsin (b). The time of simulations is 3 ns.
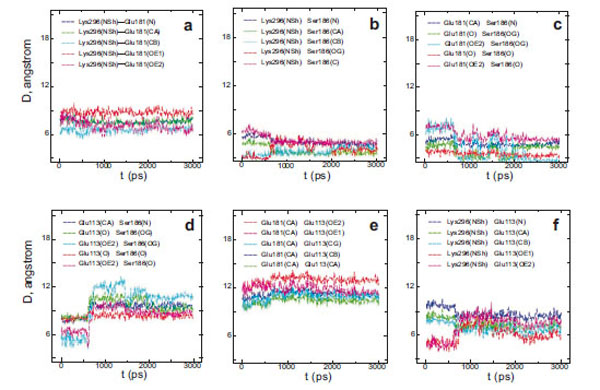
Interatomic distances (D) between different atoms of Glu181 and the PSB nitrogen (a), Ser186 and the PSB nitrogen (b), Glu181 and Ser186 (c), Glu113 and Ser186 (d), Glu113 and Glu181 (e), Glu113 and the PSB nitrogen (f) in wild rhodopsin.
From a three-dimensional image of our model, we have seen that the oxygen (ОЕ2) atoms of the carboxy groups of Glu113, Glu181, and the N atom of PSB form a triangle in which Ser186 (more precisely, its hydroxy group) is located in the segment connecting Glu181 and the N atom. We have analyzed the dynamics of Ser186. It was found that the O atom of the Ser186 hydroxy group is located at the same distance from the O atom of the Glu181 carboxy group as the N atom of the PSB group. This distance is ~3 Å (Fig. 3b and c). Presumably, a strong hydrogen bond is actually formed between Glu181 and PSB via Ser186. Thus, according to our model, Glu181 and Ser186 could make a greater contribution to PSB stabilization than Glu113 (Fig. 3a-f; Fig. 4a).
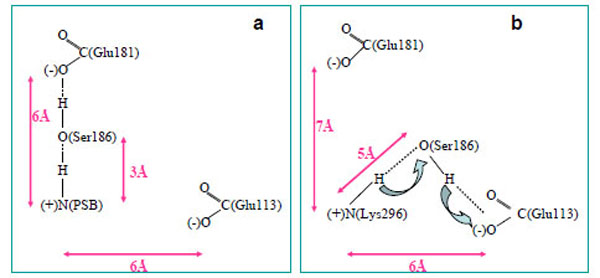
A scheme of PSB linkage stabilization in wild rhodopsin (a) and free opsin (b).
In the case of opsin, where there is no 11-cis-retinal, a hydrogen bond is not formed between Glu181 and Lys296 via Ser186. Moreover, Ser186 moves from Glu181 towards Glu113 (Fig. 4b), which undoubtedly points to rhodopsin photolysis. As light quantum is absorbed and, as a consequence, electron density redistribution in the isomerized retinal takes place [35], the electrostatic interactions of the chromophore with the protein environment are disturbed. 11-cis-retinal photoisomerization to the all-trans-configuration initiates the conformational rearrangement of the nearest amino acid residues in the chromophore centre following the disruption of the hydrogen bond between Glu181 and Ser186. As a result, Ser186 is looking for another participant to form the hydrogen bond. Glu113 is the best candidate as a proton acceptor. However, in this case, the strong hydrogen bond cannot be generated (we suppose that the minimal distance between PSB and Ser186 increases from 3 Å to 5 Å), proton from PSB can move through Ser186 to Glu113, and Schiff base linkage is hydrolyzed. Then, the phototransduction processes at the metarhodopsin II stage of photolysis are initiated in the photoreceptor cell, and the all-trans-retinal is removed from the protein.
Thus, our theoretical calculations allow us to suggest that Glu181 through Ser186 stabilize the PSB linkage in rhodopsin. Ser186 serves as a switch in the molecular mechanism which regulates PSB linkage stability. Glu113 is likely to serve in this mechanism as a proton acceptor, and then, a stabilizing factor.
The E181K Mutant Form of Rhodopsin
We have performed theoretical calculations on the E181K mutant form of rhodopsin. A molecular dynamics simulation has shown that the absence of Glu in position 181 leads to Ser186 replacement (Fig. 2b). It moves towards Glu113 as in the case of free opsin without 11-cis-retinal (Fig. 4b). We suggest that the weak hydrogen bond between the N atom of PSB and Glu113 is generated via Ser186. So, in the E181K mutant rhodopsin, a PSB proton can easily break away, promoting Schiff base hydrolysis. In other words, the PSB linkage is becoming unstable. It leads to the impairment of the native visual pigment formation and, as a result, to the initiation of photoreceptor cell pathology.
Molecular Dynamics Simulation of the Amino Acid Residues Ser334 and Ala241 in Wild and Mutant Forms of Rhodopsin
The Wild Form of Rhodopsin
It is known that amino acid residues Ser334 and Ala241 are located at the coupling place of the G-protein transducin [36,37]. In dark-adapted rhodopsin, this centre is not available to transducin. After rhodopsin photoactivation, it becomes available to transducin.
Our theoretical calculations made it clear that during a 3-ns simulation, the amino acid residue Ser334, which belongs to the C-terminal tail, and Ala241, which is of the third loop, come in a close contact in the case of wild rhodopsin (Fig. 5a). Changes in the interatomic distances of these two amino acid residues during the simulation process were assessed. The plot shows that at the initial time point of the simulation (t=0), these amino acid residues are quite far from each other (6–8 Å) and the fluctuations of these areas have a high amplitude. However, they come much closer to each other in 400 ps: the amplitude of the oscillations becomes markedly smaller and the distance between Ser334 and Ala241 becomes so short (about 3 Å) that the formation of a hydrogen bond becomes possible. Since these amino acid residues are in the active transducin binding center and since this center becomes accessible to transducin after the photoactivation of rhodopsin, we can suppose that the Ser334 and Ala241 bonding may play a role as a triggering intramolecular mechanism, which blocks transducin coupling.
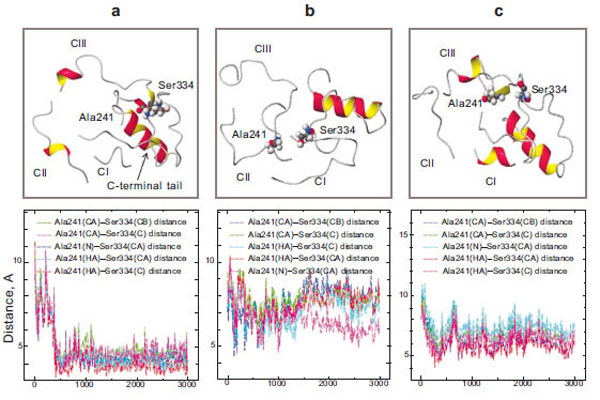
Molecular dynamics simulation of the cytoplasmic surface domain of the rhodopsin molecule: the CI, CII, CIII-loops and C-terminal tail (top) and diagrams of Ala241 – Ser334 interatomic distances (bottom) in wild rhodopsin (a), free opsin without 11-cis-rerinal (b), and the E181K mutant form (c).
Unlike the molecular dynamics of the cytoplasmic loops, that of the protein lacking 11-cis-retinal is essentially the same as at the initial stage of the simulation. It should be noted that in the absence of the 11-cis-retinal in the chromophore center, the hydrogen bond between Ser334 and Ala241 is not formed (Fig. 5b). Thus, the presence of 11-cis-retinal in opsin initiates these conformational rearrangements of the cytoplasmic loops.
The E181K Mutant Form of Rhodopsin
In the case of the mutant form, the new hydrogen bond between the amino acid residues Ser334 and Ala241 is not formed (Fig. 5c). It could be assumed that the active site in the cytoplasmic domain of the protein that is responsible for binding transducin is not completely blocked. There is a possibility of transducin interaction with the active center of rhodopsin in the absence of light. Disturbances in the interaction of these amino acid residues are believed to lead to manifestations of nonspecific activity in the II class mutant. The permanently activated state of the visual cell leads finally to its death. As a result, degenerative processes take place in the retina.
DISCUSSIONS AND CONCLUSIONS
In summary, the work presents a molecular dynamics study of rhodopsin aimed at identifying the structural determinants of the abnormal behaviour of the E181K mutant, which is associated with some variants of autosomal dominant retinitis pigmentosa. Based on MD analysis data, it is suggested that the mutant disrupts the interactions of the chromophore with its protein neighborhood, as well as causes a structural change in the cytoplasmic domain.
First of all, specially mentioned should be a methodological innovation in simulating the rhodopsin protein and tracing up its conformational properties. The main fact is that rhodopsin is a membrane protein, which was simulated in an aqueous solution. As noted above, for the rhodopsin protein we first observed its rearrangement – namely, the adaptation of the 11-cis-retinal chain inside the chromophore pocket, which takes about 0.4 ns. The most important event – 11-cis-retinal rearrangement (adaptation) inside the chromophore center of the rhodopsin – thus occurs quickly, and extending rhodopsin relaxation further beyond a 3-ns range for the dark-adapted state would not provide any more conformational features. Though our model is a protein molecule in an aqueous environment (rhodopsin is a membrane protein), the rhodopsin dynamics described in this paper is, nevertheless, in a good correlation with experimental [28] and theoretical studies, including those with models in lipid environments [14,29,38,39]. This allows one to extrapolate the simulation results to the in vivo dark physiological regeneration of rhodopsin. Total rhodopsin conformation in an aqueous water solution at the studied time scale of 11-cis-retinal adaptation is similar to the behavior of the rhodopsin molecule embedded in a lipid bilayer.
The main event – 11-cis-retinal adaptation – determines all subsequent conformational events of rhodopin conformation. Our MD simulation results, with regard to the conformational shape of 11-cis-retinal, correlate with a number of experimental observations and quantum chemistry calculations. Not only does chromophore “adaptation” in rhodopsin’s binding pocket bring the rhodopsin molecule to a state of “high alert”, but it also stabilizes the rhodopsin inactive conformation as a G-protein-coupled receptor. It is well known that 11-cis-retinal in dark-state rhodopsin has a non-planar, distorted, and twisted conformation [28,40]. This makes its conformational state different from that of the retinal protonated Schiff base in a solution [39]. However, the molecular mechanisms that “force” the chromophore to assume such an energetically “unfavorable” configuration are still unclear. The role of the protein environment in the formation of this configuration of 11-cis-retinal in rhodopsin is now becoming clear. This is the key point of the present simulations, where we first build a real-time model of 11-cis-retinal adaptation in opsin. Based on animated pictures and over 3000-picosecond calculations of interatomic distances, torsional rotation angles, etc., we describe now the model of the dark “adaptation” of retinal incorporated in opsin. The model describes well the origin of the beta-ionone ring twist, resulting in polyene chain rotation, which is assumed to explain that 11-cis-retinal exists in such a distorted conformation that makes it stable, but at the same time ready for efficient ultrafast photoisomerization. The results on the rotational angles of methyl groups C19 and C20 are in good agreement with experimental NMR-spectroscopic data [41], where the deviation angles of the C9-C19 and C13-C20 vectors from the axis perpendicular to the photoreceptor membrane are approximately 42° and 30°, respectively. Thus, our simulation results on chromophore dynamics seem to correlate well with both experiments [3,15,28,40,42,43] and theoretical quantum chemistry calculations [14,38,44]. This clearly demonstrates that retinal in dark rhodopsin has a twisted configuration.
Our calculations also demonstrated a high level of ordering for the fragments of N-terminal polypeptide chain and intradisc loops compared to the cytoplasmic domain, which is well correlated with the X-ray diffraction data [15,40]. This is also supports the well known views on the important role of the intradisc domain in stabilization of the molecule in the photoreceptor membrane [45-47]. Based on the details of the 11-cis-retinal adaptation process, we have expanded the scope of the rhodopsin model to include the mutated version.
Thus, in the case of the E181K point mutation, there is an electrostatic interaction disturbance between the 11-cis-retinal chromophore and the surrounding amino acid residues within the chromophore centre of the rhodopsin molecule. As a result, the stable Schiff base linkage cannot be generated, and the rhodopsin molecule cannot be formed. The disturbance of the electrostatic interactions of the 11-cis-retinal chromophore with the surrounding amino acid residues leads to disorder in cytoplasmic loop packing. Consequently, mutant rhodopsin could be permanently activated, which leads to cell death. The impairment of PSB stability in mutant rhodopsin forms was also confirmed by other theoretical data [48].
It was attempted to find inhibitors of mutant rhodopsin that would be able to inhibit the visual pigment both in the dark and light. Some modified forms of the 11-cis-retinal chromophore were applied to get stable PSB [49–51]. However, the medicine has not been found. Our theoretical calculations show that even if the stable PSB in such mutant form of rhodopsin was obtained, there would be no guaranty of coupling place blocking for the G-protein transducin. In the near future, of retinal gene therapy might solve the problem of retinitis pigmentosa – an inherited degenerative disease of the retina [52].
CONFLICT OF INTEREST
The author(s) confirm that this article content has no conflicts of interest.
ACKNOWLEDGEMENTS
This work was performed using computer facilities of the Computational Astrophysics Laboratory (CAL) at the Institute of Physical and Chemical Research (RIKEN), Japan. The authors are grateful to CAL for providing computer time and access to a supercomputer and the MDGRAPE-2 special-purpose computer for performing the molecular dynamics calculations of protein molecules, and to the CAL Director T. Ebisuzaki for the support of licensed software and MDGRAPE-2 computers for protein modeling. The authors would like to specially thank Mr. Sergei Negovelov (JINR) for technical assistance and helpful comments.
REFERENCES
[PubMed Link]
[PubMed Link]

