All published articles of this journal are available on ScienceDirect.

Biochemical and Computational Approaches to Improve the Clinical Treatment of Dopa Decarboxylase-Related Diseases: An Overview
Authors Info & Affiliations
Abstract
Dopa decarboxylase (DDC) is a pyridoxal 5’-phosphate (PLP)-dependent enzyme that by catalyzing the decarboxylation of L-Dopa and L-5-hydroxytryptophan produces the neurotransmitters dopamine and serotonin. The functional properties of pig kidney and human DDC enzymes have been extensively characterized, and the crystal structure of the enzyme in the holo- and apo-forms has been elucidated. DDC is a clinically relevant enzyme since it is involved in Parkinson’s disease (PD) and in aromatic amino acid decarboxylase (AADC) deficiency. PD, a chronic progressive neurological disorder characterized by tremor, bradykinesia, rigidity and postural instability, results from the degeneration of dopamine-producing cells in the substantia nigra of the brain. On the other hand, AADC deficiency is a rare debilitating recessive genetic disorder due to mutations in AADC gene leading to the inability to synthesize dopamine and serotonin. Development delay, abnormal movements, oculogyric crises and vegetative symptoms characterize this severe neurometabolic disease. This article is an up to date review of the therapies currently used in the treatment of PD and AADC deficiency as well as of the recent findings that, on one hand provide precious guidelines for the drug development process necessary to PD therapy, and, on the other, suggest an aimed therapeutic approach based on the elucidation of the molecular defects of each variant associated with AADC deficiency.
INTRODUCTION
Pyridoxal 5’-phosphate (PLP)-dependent enzymes, except glycogen phosphorylase, synthesize, degrade and inter-convert amino acids. They catalyze a wide variety of reactions including racemisation [1], transamination [2], decarboxylation [3], α-elimination [4], β-replacement [5] and γ-replacement [6]. They play indispensable roles in the primary metabolism of cells as well as in secondary metabolism, synthesizing important signaling molecules. Consistent with these metabolic roles, inhibitors of many PLP-enzymes have been and/or are used against certain types of neurological affections [7, 8], infectious diseases [9-11], and cancer [12]. The main hindrance in designing a PLP-enzyme inhibitor is the common chemistry of this family leading to low specificity and broad action. B6 enzyme inhibitors in medicinal use are substrate analogs but they follow different mechanisms for inhibiting their target enzymes. In fact, vigabatrin, difluoromethylornithine and D-cycloserine act as suicide inhibitors of human γ-aminotransferase [11], ornithine decarboxylase [12] and alanine racemase [11], respectively, while pregablin competitively inhibits human γ-aminotransferase [8], and benserazide and carbidopa irreversibly bind Dopa decarboxylase (DDC), thus blocking its active site. Recently, a general procedure for designing phosphopyridoxyl substrate-conjugate inhibitors of PLP-enzymes modified to make them membrane-permeable has been described [7]. Favorable targets for this approach appear to be enzymes that have relatively low PLP affinity or undergo transamination as a significant side reaction with their amino acid substrate, thus forming the non covalently, less firmly bound pyridoxamine 5’-phosphate form of the cofactor.
Moreover, some PLP-enzymes are pharmacotherapeutic targets since their deficit is responsible for rare diseases, a heterogeneous group of disorders that encompass more than 14 distinct metabolic diseases. The incidence as a group is about 1:10000, but, taken individually, each disease is very rare and many have an incidence of less than 1:1000000. The list of these rare disorders along with the symptoms and the enzyme involved is reported in Table 1. The molecular basis of many of these diseases has been analyzed by comparing the characteristics of the normal enzyme with those of the pathogenic variants either in their recombinant purified form [13-17] or expressed in cellular systems [18, 19]. A particular emphasis has been addressed to pathogenic variants of human peroxisomal liver alanine:glyoxylate aminotransferase [20-26] and cysthathionine- β-synthase [27-29]. It should be also mentioned that several neurological pathologies such as autism, schizophrenia, Alzheimer’s disease, epilepsy and Down’s syndrome can be caused by the deficiency of PLP, often determined by a reduced activity of PLP salvage pathway enzymes including pyridoxal kinase and pyridoxine 5’-phosphate oxidase [30-32].
Rare Diseases Involving PLP-enzymes
| Rare Disease | Involved Enzyme | Main Symptoms |
|---|---|---|
| Aromatic L-amino acid decarboxylase deficiency [61] | DOPA decarboxylase | delay in development, abnormal movements, oculogyric crisies, vegetative symptoms |
| Cystathioninuria [69] | Cystathionine-γ-lyase | abnormal urinary excretion of cystathionine, fibrotic liver |
| GABA-transaminase deficiency [70] | γ-aminobutyric acid-transaminase | hyperreflexia, hypotonia, lethargia, macrosomia, mental retardation, and siezures |
| Gilles de la tourette syndrome [71] | Histidine decarboxylase | arm thrusting, eye blinking, repeated throat clearing or sniffing, shoulder shrugging |
| Glycine encephalopathy [72] | P protein (a pyridoxal phosphate-dependent glycine decarboxylase) | mental retardation, hypotonia, seizures, brain malformations, ataxia |
| Hereditary sensory and autonomic neuropathy Type I [73] | Serine palmitoyltransferase | sensory deficit in the distal portion of the lower extremities, chronic perforating ulcerations of the feet and progressive destruction of underlying bones, sweating |
| Homocystinuria [74] | Cystathionina-β-syntase | high level of homocysteine, endothelial injury, isk of other artery or vein diseases |
| Ornithine aminotransferase deficiency (Gyrate atrophy) [75] | Ornithine aminotransferase | poor vision at night or in dim light, peripheral vision loss |
| Primary hyperoxaluria Type I [76] | Alanine:glyoxylate aminotransferase | oxalosis, nephrocalcinosis urolithiasis, nephrolithiasis, kidney stones |
| Smith-Magenis syndrome [77] | Serine hydroxymethyltransferase | square-shaped face with deep-set eyes, engaging personalities, short stature, scoliosis, reduced sensitivity to pain and temperature, and a hoarse voice |
| Stiff-Man Syndrome [78] | Glutamic Acid Decarboxylase | muscle rigidity that waxes and wanes with concurrent spasms, encephalomyelitis, epilepsy, cerebral palsy, or cerebellar deficits |
| Type II Tyrosinemia (Richner-Hanhart syn-drome) [79] | Tyrosine aminotransferase | excessive tearing, photophobia, eye pain and redness, and painful skin lesions on the palms and soles, mental retardation |
| Xanthurenic aciduria [80] | L-Kynurenine hydrolase | vomiting, jaundice, high excretion of Xanthurenic acid |
| "X-linked" sideroblastic anemia [81] | δ-aminolevulinate synthase | microcytic red blood cells hypochromic an abnormal accumulation of iron in red blood cells |
Dopa decarboxylase (DDC), a PLP-enzyme responsible for the synthesis of the neurotransmitters dopamine and serotonin, represents a double-face therapeutic target because it is involved both in Parkinson’s disease (PD) and in a rare neurometabolic disorder named aromatic amino acid decarboxylase (AADC) deficiency. In this review the attention is focused on the structural elements and functional features of DDC, on the current DDC-directed therapies for PD and AADC deficiency as well as on recent findings providing important guidelines for the future management of patients affected by these diseases.
DDC: STRUCTURAL AND FUNCTIONAL PROPERTIES
DDC, a PLP-dependent homodimeric enzyme, is an α-decarboxylase that catalyzes the conversion of L-aromatic amino acids into their corresponding amines. DDC, abundant in the nervous system and kidney, is central in the synthesis of dopamine and serotonin. Thus, its essential function is to provide the organism with these biogenic amines. Pig kidney, rat liver and human DDC enzymes have been cloned, expressed in E. coli and purified [16, 33, 34]. Our group has been involved for long time in the characterization of the pig kidney enzyme [10, 35-42], and recently in that of human DDC [16]. Pig kidney DDC and human DDC display similar spectroscopic features, kinetic parameters, susceptibility to proteolysis and thermostability [16]. The resolution of the three-dimensional structure of holoDDC in its ligand-free form and in complex with the anti-Parkinson drug carbidopa has allowed to point out that the structure is a tightly associated dimer in which the active site is located near the monomer-monomer interface, although being mainly composed of residues from one monomer. Moreover, thanks to solved structures, we were able to identify the residues involved in the binding of the coenzyme, to evidence the antiparkinsonian drug binding mode, and to suggest which amino acid residues may be involved in catalysis [43]. Each of the two monomers is composed of three distinct domains. The large domain contains the PLP binding site and includes a core formed by a seven-stranded mixed β-sheet surrounded by eight α-helices in a typical α/β fold. The C-terminal small domain comprises a four-stranded antiparallel β-sheet with three helices packed against the face opposite to the large domain. The N-terminal domain, composed of two parallel helices linked by an extended strand, lies like a flap over the top of the second subunit. Until now, only few studies have been published that describe the functional role of the N-terminus of PLP-enzymes [44-46]. For human DDC, the recent resolution of the crystal structure of the enzyme in its apo form has provided evidence for an unexpected open bivalve conformation (as compared with the holoenzyme) with the interface between the N-terminal domains functioning as the hinge [47], thus suggesting a role for the N-terminus in the formation of the closed dimeric conformation of the holoenzyme.
PD
PD is a neurodegenerative disorder characterized by tremor, bradykinesia, rigidity, and postural instability. These symptoms are caused by the reduced levels of dopamine, consequent to the progressive degeneration of dopamine-producing cells in the substantia nigra of the brain [48]. Dopamine itself cannot pass the blood-brain barrier and, therefore, cannot be used as a drug in the PD treatment. Thus, one of the most effective clinical treatments of the disease is to routinely administer L-Dopa, in combination with a DDC inhibitor. The rational of this treatment is that since L-Dopa is rapidly converted to dopamine in the blood stream, only a small percentage of a given dose of Dopa, when administered as a drug, can reach the nervous system (Scheme 1A). By adding a DDC inhibitor, greater amounts of L-Dopa can reach the brain (Scheme 1B), where the level of dopamine will result to be substantially increased. The most commonly DDC inhibitors used in the treatment of PD are carbidopa ((2S)-3-(3,4-dihydroxyphenyl)-2-hydrazino-2-methylpropanoic acid; MK485) and benserazide ((RS)-2-amino-3-hydroxy-N′-(2,3,4-trihydroxybenzyl)propanehydrazide; Ro-4-4602). In animals and humans benserazide is completely metabolized to serine and trihydroxybenzylhydrazine (Ro-4-5127) before it reaches the arterial blood [49]. Thus, it is likely that trihydroxybenzylhydrazine represents the actual DDC inhibitor. Indeed, while benserazide is not a powerful DDC inhibitor [50], carbidopa and trihydroxybenzylhydrazine, both substrate analogs endowed with a substituted hydrazine function, have been found to bind pig kidney DDC by forming a hydrazone linkage with PLP, and work as powerful irreversible DDC inhibitors [50, 51]. However, several side effects (nausea, hypotension, arrhythmias, gastrointestinal bleeding and serious psychiatric symptoms) ensue from co-administration of L-Dopa with carbidopa or benserazide [52, 53], which are possibly related to the following findings: (i) carbidopa and trihydroxybenzylhydrazine react non-enzymatically with free PLP [52-54], (ii) the condensation products formed between PLP and hydrazine derivatives are the most potent inhibitors of pyridoxal kinase, an enzyme involved in PLP biosynthesis [55, 56], and (iii) high levels of homocysteine inversely correlate with PLP concentrations in the treated patients [57-59]. Thus, the current protocol seems to alter the PLP metabolism. Recently, a promising step forward in the drug development for PD has been accomplished. A virtual screening protocol, integrating pharmacofore search and molecular docking, has been developed starting from the atomic structure of the DDC-carbidopa complex [43]. The combination of these computational methods with enzymatic assays and spectroscopic analyses has provided ten compounds identified as competitive inhibitors with Ki values in the low micromolar range and predicted to not cross the blood-brain barrier [60]. This approach, the first reported in the field of DDC inhibitors discovery, has represented an excellent staring point to further hit-to-lead development. In fact, it is relevant that these molecules, unlike carbidopa and trihydroxybenzylhydrazine, are devoid of an hydrazinic group, do not bind free PLP and act as good reversible inhibitors. Compounds endowed with such chemical features are more selective than the drugs commonly used in the treatment of PD. Thus, when improved and developed as drugs, they could reduce the very undesirable side effects of the current therapeutic strategy. On the basis of the above considerations and results, the drug development for the PD therapy seems to be a promising endeavor.
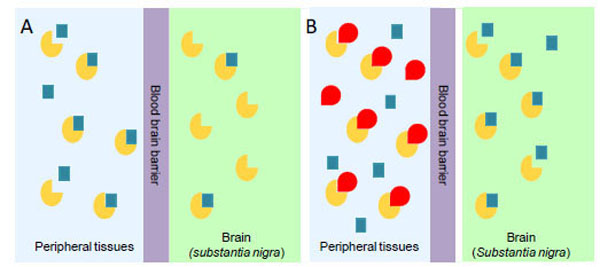
Availability of administered L-Dopa in the substantia nigra of brain in the absence (A) or in the presence (B) of an inhibitor of peripheral DDC. The yellow and the red symbols represent DDC and DDC inhibitor, respectively; the blue squares represent L-Dopa.
AADC DEFICIENCY
Recessive inherited deficiency of DDC (OMIM#608643) results in a severe neurometabolic disorder with developmental delay, abnormal movements, oculogyric crises and vegetative symptoms. Since the first report in 1990 [61], about 50 cases of DDC deficiency have been reported [62]. Patients display a typical pattern characterized by a distinct reduction of the stable degradation products of dopamine and serotonin, homovanillic acid and 5-hydroxyindolacetic acid, accompanied by the increase of the level of the precursors of dopamine and serotonin, L-Dopa and 5-hydroxytryptophan, respectively. Additionally, the levels of 3-ortho-methyldopa, resulting from methylation of accumulating L-Dopa, are clearly increased [63]. The diagnosis is confirmed by measuring the enzyme activity of DDC in plasma or cells [64]. Although about 30 mutations on the AADC gene have been identified and much is known about the clinical phenotype of the patients, the enzymatic phenotypes are so far largely unknown as well as of course the molecular mechanisms by which each mutation leads to the deficiency of DDC. While the effect of nonsense and frameshift mutations that lead to the complete loss of the gene product is straightforward, single amino acid substitutions leading to the synthesis of an aberrant gene product can in principle affect the enzymatic activity, the binding of the coenzyme, the protein stability and /or folding, the enzyme half-life inside the cell, etc. Moreover, it should be emphasized that many patients affected by DDC deficiency are compound heterozygous and their enzymatic phenotype depends on the combination of the effect of one mutation on a monomer and a different mutation on the other monomer. The core treatments for AADC deficiency are pyridoxine, MAO-B inhibitors, and dopamine agonists. Administered pyridoxine is phosphorylated by pyridoxal kinase and then subsequently converted to PLP by pyridoxal oxidase. The aim of this treatment is to provide an excess of PLP, which may in turn bolster or enhance residual DDC activity. MAO-B inhibitors (tranylcypromine, selegiline, moclobemide) are given to prevent the breakdown of the limited amounts of dopamine and serotonin produced by AADC deficient patients. Dopamine agonists (bromocriptine, pergolide, pramipexole and ropinirole) are administered in AADC deficiency in an attempt to stimulate dopamine receptors and consequently replace dopamine neurotransmission (Scheme 2) [65]. It should be mentioned that in the last years a gene therapy approach has been explored, and good indications have been provided for the potential success of this technique for AADC deficiency [65, 66]. The response to the therapies in clinical use has been variable, but the overall outcome remains poor, probably reflecting the allelic heterogeneity. Moreover, since drugs were almost always given in combination, the response is hard to be ascribed to a single drug. Again, these classic treatments tend to address the more distal aspects of the disease process, i.e. symptoms rather than causes. A very important issue for the diagnosis and the therapy of diseases correlated to protein malfunction is the understanding of the multiple mechanisms that relate the specific mutants with the pathology. In fact, the knowledge of the enzymatic phenotype associated with each mutation represents the basis for predicting the response of patients to the available therapies as well as to project new therapeutic strategies. The structural and/or functional effect(s) that amino acid substitutions produce on DDC has been recently investigated. A first approach in this direction is the analysis of the crystal structure of the enzyme to rationalize and interpret the impact of some disease-specific mutations in terms of their likely effects on enzyme’s tertiary and quaternary structure. Fig. (1) represents an overview of DDC pathogenic mutation sites. Visual inspection of the three-dimensional structure of DDC in its holo-form suggests that, despite the variability in the space distribution, some physico-chemical effects shared by the disease-associated mutations might lead to similar enzymatic phenotypes. In particular, some point mutations (S147R, G102S, F309L, A275T, and R347Q) may affect the integrity of the active site, some (L38P and A110Q) might affect the stability of the dimer interface, while some (P47H and L38P) are expected to mostly compromise the stability of the monomeric form of the enzyme. For a number of mutants (R358H, V460G, G123G, S250F, R285W) the substitution is expected to perturb important hydrophobic patches within the structure, which might lead to misfolding or protein aggregation in cases where the perturbation occurs in solvent-exposed areas. Three mutants (L408I, R412W, R447H) are located in the small C-terminal domain in sites neither involved in the dimerization, nor in the catalytic areas. Such a “hot spot” in a well-defined domain might have functional consequences, which are currently unknown. Finally, the resolution of the structure of apoDDC [47] has highlighted that some pathogenic mutations (such as Y79C, H70T, H72Y and T69M) (Fig. 2) involve residues important for the conformational changes occurring upon PLP binding, thus possibly affecting the apo-to-holo transition.
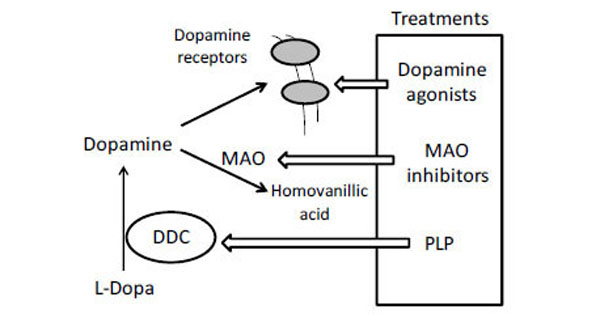
Current clinical management of AADC deficiency.
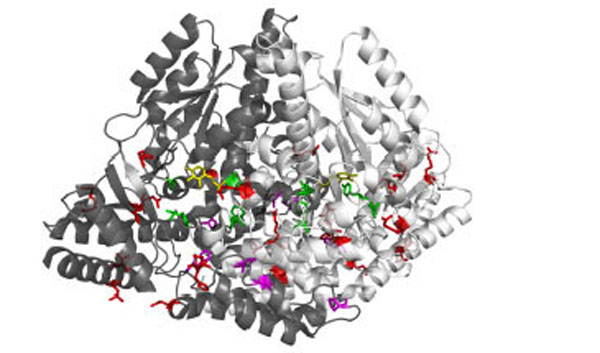
3D representation of the dimeric DDC molecule in which the two chains are represented as white and grey ribbon respectively. Sites of mutation are sorted in: mutations near the active site (green sticks), mutations at the dimer interface (magenta sticks), and mutations in the peripheral regions (red sticks). PLP molecules are also highlighted as yellow sticks.
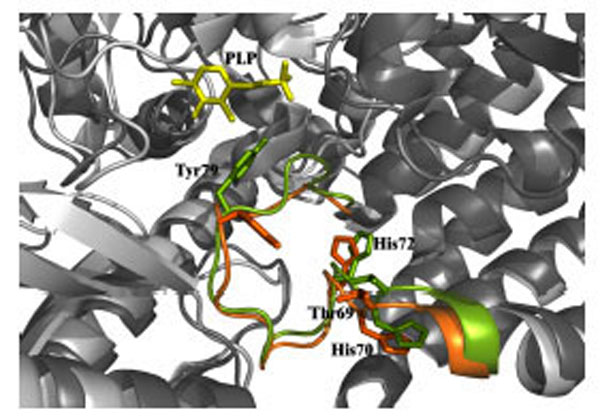
Structural superposition of the apo (green) and holo (orange) DDC active site. Thr69, His70, His72 and Tyr79 residues are represented as stick. PLP molecule is represented as yellow stick.
Recently, biochemical and bioinformatic investigations of several pathogenic DDC variants in the recombinant purified form have been instrumental to uncover their molecular defects and to suggest an appropriate therapeutic strategy for AADC deficiency patients. The G102S, F309L, S147R and A275T variants associated with AADC deficiency, in which mutations concern amino acid residues interacting directly or indirectly with PLP and/or its microenvironment, have been constructed, expressed and purified [16]. These mutations cause, even if to different extents, a reduced PLP binding affinity (in the range 1.4-170-fold), an altered PLP binding mode, and, except for the S147R mutation, an increased Km value for L-Dopa (in the range 3-35-fold). Moreover, all the variants display a reduced kcat value, which appears to be correlated to a mispositioning of the external aldimine at the active site. In fact, in comparison with wild-type DDC, the external aldimines formed by the variants with L-aromatic amino acids exhibit different absorbance and dichroic features, do no protect against tryptic limited proteolysis, and, with time, in addition to aromatic amines production, lead to a decrease in the original PLP content and a concomitant increase in cyclic-PLP-substrate adducts. On these bases, it has been suggested that the examined pathogenic mutations are responsible for an active site perturbation, and, consequently substrate binding does not permit to completely reach a conformation where the catalytic groups are in correct proximity and orientation to allow a high level of reaction specificity, i.e., a conformation productive for decarboxylation. Such a conformation is, instead, achieved in wild-type DDC upon substrate-induced conformational change [67].
These data have allowed not only to understand the molecular defects of the examined DDC pathogenic variants, but also to give some suggestions for the treatment strategy of AADC deficiency patients bearing these mutations. In particular, a therapy consistent in pyridoxine plus L-Dopa and L-Dopa alone has been proposed for patients carrying F309L and G102S mutations, respectively. It is interesting that Chang YT. et al., [68] have observed the effectiveness of L-Dopa administration to three siblings with the G102S mutation. Although up to date, no patients with homozygous genotype harboring S147R or A275T mutations have been identified, but only compound heterozygous patients [68], it has been proposed that pyridoxine treatment could effectively counteract the effects of the A275T mutation. No similar conclusion can be drawn for the S147R variant whose catalytic activity is 840-fold lower than that that of the wild-type, even in the presence of saturating PLP concentration.
Therefore, it appears highly desirable to extend this approach at the protein level to as many as possible other mutations associated with AADC deficiency in order to (i) understand the molecular basis of the pathogenicity of each mutation associated with the disease, (ii) advance proposals for the different drugs to overcome the disease, and (iii) help in listing patients in different therapeutic categories depending on the molecular defect(s) of each mutation. Advances in the understanding of the molecular etiology of AADC deficiency shift the attention towards the more proximal aspects of the disease process, i.e. the causes rather than the symptoms. This approach at the protein level could open new possibilities to (i) rationalize the choice of pharmacological agents to counteract the adverse effects of these mutations and (ii) prevent loss of time and unsuitable treatment regimens biochemically futile.
CONCLUSIONS
DDC, a PLP-dependent enzyme, plays a key role in supplying the organisms with the neurotransmitters dopamine and serotonin. The enzyme is a pharmacotherapeutic target since it is involved in PD, a neurodegenerative disorder, and in AADC deficiency, an inherited rare neurometabolic disease. In this review, we point out how biochemical and computational findings could be instrumental for improving the current treatments of these pathologies.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflicts of interest.
ACKNOWLEDGEMENTS
None declared
REFERENCES
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link] [PMC Link]
[PubMed Link] [PMC Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]

