All published articles of this journal are available on ScienceDirect.

Thalidomide Analogues Suppress Lipopolysaccharide-Induced Synthesis of TNF-α and Nitrite, an Intermediate of Nitric Oxide, in a Cellular Model of Inflammation
Authors Info & Affiliations
Abstract
An unregulated neuroinflammation accompanies numerous chronic and acute neurodegenerative disorders and it is postulated that such a neuroinflammatory component likely exacerbates disease progression. A key player in brain inflammation is the microglial cell; a vital soluble factor synthesized by activated microglial cells is the key cytokine, tumor necrosis factor–alpha (TNF-α). Additionally, microglial cells release IL-1α/β, reactive oxygen species (ROS), such as superoxide (O2-) and reactive nitrogen species (RNS) like nitric oxide (NO). Nitric oxide reactive oxygen species can undergo various forms of interactions in cells whereby the synthesis of RNS / ROS intermediates are generated that can damage cell membranes. The presence of oxidative damaged cells is implicated with the abnormal cellular activity in brain or in the spinal cord, and is a classical feature of neurodegenerative disorders. To aid characterize this process, a quantitative analysis of nitrite generation was undertaken on agents developed to lower TNF-α levels in cell culture. Nitrite is a stable end product of nitric oxide metabolism and, thereby, acts as a surrogate measure of the highly unstable nitric oxide. Utilizing a RAW 264.7 cellular model of lipopolysaccharide-induced inflammation that induces high levels of TNF-α protein accompanied by a robust generation of nitrite, the properties of a series of thalidomide-based TNF-α synthesis inhibitors were evaluated to reduce the levels of both. Specific analogues of thalidomide effectively suppressed the generation of both TNF-α and nitrite at well-tolerated doses.
INTRODUCTION
Alzheimer's disease (AD) is one of the most prevalent neurological disorders in humans and, according to recent estimates, there are in excess of 27 million people worldwide who have been diagnosed with the condition, of which 5 million reside in North America. Based on data from ongoing studies, these numbers are anticipated to increase sharply in the near future [1]. Strongly associated with several neurological disorders, which include AD, Parkinson’s disease (PD) and amyotrophic lateral sclerosis (ALS) is the phenomenon of neuroinflammation. It is believed that, whereas the specific causes of these conditions remain to be fully elucidated, neuroinflammation plays a critical role in the neurodegeneration continuum [2-4].
In brain, a key cellular player involved in the immune response is the brain resident macrophage, the microglial cell; another principle cell is the astrocyte. Typically, microglial cells are involved in maintaining the homeostasis of the brain microenvironment. These act primarily by augmenting the clearance of cellular debris from dead or dying cells or responding to pathogens and cytokines produced by other immune cells [5, 6]. During normal conditions, microglial cells are described as ‘ramified’ or ‘resting’, wherein they are essentially dormant, yet standing by should they be needed to rapidly respond to any form of stimulation. Under potentially pathological conditions, however, these resting cells can be induced by exogenous and endogenous factors that trigger morphological changes which move the cells towards an amoeboid, activated morphology. In this activated state, a classical feature is the generation of an oxidative burst, or respiratory burst, wherein the cells are actively responding to, and synthesizing an array of soluble factors. These cellular factors, if unregulated, may induce neuronal dysfunction and degeneration, or they may transform additional resting microglial / astrocyte cells into activated immune cells. Such factors include the following: TNF-α, IL-1α/β, reactive nitrogen species (RNS), like nitric oxide (NO), and reactive oxygen species (ROS), such as superoxide (O2-). With regard to soluble factors released by activated microglial cells, one of the most important proteins is TNF-α [7, 8]. TNF-α signaling can be mediated via several pathways. (1) Through the c-Jun N-terminal kinase pathway that, depending on which transcription factors are activated, may lead to cell death or cell survival effects [9-6]. (2) Via the activation of a caspase-dependent signaling pathway, which may cause apoptotic cell death [17]. (3) Additionally, by TNF-α activation of the transcription factor NFκB, which, in turn, induces the activation of survival pathways [18]. Hence, TNF-α signaling can be associated with both cell death and cell survival properties and, consequently, this makes a principle role for TNF-α difficult to define for a given set of cellular circumstances. Perhaps a key feature of this potent pro-inflammatory cytokine is related to the apparent ease with which it can activate resting immune cells. This ability creates an opportunity to allow the formation of a feedback loop of TNF-α–induced unregulated neuroinflammation [19]. The potential for a feedback phenomenon becomes of major relevance in several CNS diseases when one considers that in the majority of these medical conditions altered cellular states exist in the brain which generate an ideal environment for immune cell self-activation, as exemplified by amyloid-beta peptide (Aβ) in AD.
In AD a hallmark feature, is the formation of the toxic peptide Aβ that is generated by the proteolytic actions of both β- and γ-secretase on amyloid precursor protein (APP) [20]. Both cell culture and animal studies have shown that Aβ-peptide can induce the activation of microglial cells, following which they can release cytokines and cytotoxic factors [21, 22]. Likewise in PD, a cardinal feature is the presence of α-synuclein (α-syn) protein deposits in the brain that, in a manner reminiscent of AD, can form α-syn aggregates [23] and induce the activation of microglia [23-26]. Lastly, in a subset of ALS patients bearing superoxide dismutase (SOD) mutations, defective communication between neurons and microglial cells instigates an inappropriately exaggerated cytokine release that is capable of inducing both ROS and RNS mediated damage to neurons within the spinal cord [27-32]. In each scenario, the self-activation feature of brain glial cells provides a mechanism, when it goes awry, that underpins the induction of an unregulated neuroinflammatory response [33, 34]. Accordingly, it comes as no surprise that elevated levels of TNF-α protein or the detection of TNF-α gene transcripts are associated with clinical cases of CNS disorders [2-4, 19]. Interventions aimed at limiting the biological signaling consequences of TNF-α may therefore be beneficial in such conditions.
This has been shown to be the case in peripheral immune related diseases, epitomized by rheumatoid arthritis, Crohn’s disease and psoriasis. A successful therapeutic treatment approach for these has been the use of protein-based agents to sequester free soluble TNF-α protein thereby eliminating the opportunity for the cytokine to activate its receptors. In relation to neurological diseases, a similar approach has been taken in proof of concept clinical studies involving AD patients, where benefits in specific cognitive features were detected shortly after perispinal administration of the TNF-α sequestering agent, Enbrel (Etanercept), followed by Trendelenburg positioning [35, 36]. Agents of this class, including Remicade (Infliximab) are large proteins that clear released soluble TNF-α before it can bind to its receptors; thereby, preventing TNF-α signaling. Unfortunately such protein therapeutics possess limited blood brain-barrier penetrability, making their wide utility for neurological disorders less than optimal.
An alternative approach is to limit the rate of TNF-α protein synthesis by interfering with the transcription or translation of its gene products. This is achievable through the use of small drug agents, such as thalidomide (N-α-phthalimidoglutarimide). Thalidomide possesses appropriate physicochemical characteristics (C log D value) to support high blood brain-barrier penetration and brain delivery [37] and, by reducing the half-life on TNF-α mRNA stability, it thereby lowers biosynthesis of this potent cytokine [38]. Thalidomide additionally provides a useful pharmacophore to support the synthesis and development of feasibly more potent and better-tolerated agents. The analogue, lenalidomide (Revlimid), represents an example found effective in the treatment of multiple myeloma [39, 40], and sulfur analogues appear to be particularly effective in lowering TNF-α [41,42] and may hence have a utility in the treatment of neurodegenerative diseases [2].
Extending the development of a cellular model of inflammation with utility in identifying novel thalidomide based TNF-α agents [43], herein we describe the use of this model to elucidate the actions of compounds on TNF-α and an end stage product of nitric oxide metabolism, nitrite (NO2-) [44]. Gaining insight to the relationship between TNF-α and NO has become increasingly important consequent to the indication that induction of a TNF-α response, alone, is insufficient to cause neuronal cell death. Occurrence of a secondary condition may be required and, in this regard, nitric oxide generation may play a vital role in causing TNF-α mediated neurotoxicity [45]. The present study describes the use of a cellular mouse RAW 264.7 cell model of inflammation to examine the relationship between LPS induced synthesis of TNF-α protein and nitrite, a surrogate of nitric oxide generation. This allowed analysis of the actions of agents with TNF-α and nitric oxide lowering actions. Accordingly, levels of TNF-α protein were quantified by use of an ELISA specific for mouse TNF-α and, to indirectly assess levels of nitric oxide generated by a RAW cell oxidative burst, the Griess Reagent System was utilized to measure nitrite (NO2-) [44, 46]. Selected thalidomide analogues were able to effectively suppress the synthesis of both TNF-α and nitrite more effectively than thalidomide.
MATERIALS AND METHODS
Cellular Proliferation, Nitrite and TNF-α Protein Level Quantification
The CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega, Madison, WI) is routinely used as an assay of cell proliferation, and was used herein, according to the manufacturer’s recommendations. Changes in cellular health status are determined by use of indirect measures related to the formation of a colored tetrazolium dye product that can be measured spectrophotometrically at 490 nm λ. An elevation in absorbance is indicative of an increase in cell number and, hence, cellular proliferation. Optical densities (expressed as O.D.s) were measured after 1 to 2 hours incubation. Nitrite levels in the culture media were measured by use of the Griess Reagent System (Promega, Madison, WI), following the manufacturer’s protocol. The O.D. of unknown samples was read at 520 nm λ, compared to a sodium nitrite standard curve (1.5 µM to 100 ìM) and nitrite measured media concentrations expressed as µM units. As the lowest nitrite concentration on the Griess Reagent System standard curve was 1.5 µM, this was chosen as the effective cutoff for defining measurable nitrite concentrations. TNF-α protein levels were measured by use of an ELISA specific for mouse TNF-α protein (BioLegend, San Diego, CA) and are expressed as a % change from their appropriate control or as pg/ml.
RAW 264.7 Cell Culture
RAW 264.7 cells derived from ATCC (Manassas, VA, USA) were grown in DMEM media supplemented with 10% FCS, penicillin 100 µ/ml and streptomycin 100 ìg/ml, and maintained at 37°C and 5% CO2. Cells (100 or 200 x 103) were seeded in 24 well plates and, 24 hours later, were utilized in studies. Two hours prior to the initiation of any study, the seeding media was replaced with fresh media (1 ml), and the cells were allowed to equilibrate at 37°C and 5% CO2.
Lipopolysaccharide-Induced Changes in RAW 264.7 TNF-α, Nitrite Levels and Cell Viability
RAW 264.7 cells were challenged with LPS (Sigma, St Louis, MO: serotype 055:B5) from 0.1 ng/ml to 200 ng/ml. Each concentration was prepared as a 100 times stock solution in sterile saline and applied directly to individual wells in a 24 well plate. At 24 and 48 hours following the addition of LPS, conditioned media was harvested and analyzed for quantification of secreted TNF-α protein and nitrite levels. Fresh media was replaced into the wells and cell viability was then assessed.
Thalidomide and Analogues
Thalidomide and analogues were prepared in tissue culture grade dimethylsulphoxide (DMSO, Sigma). RAW 264.7 cells were pretreated with thalidomide, analogues or vehicle (at 1:200 dilution in 100% DMSO) one hour prior to LPS challenge. The effects of thalidomide and four thiothalidomide analogues (Fig. 1) were assessed at 10 μM and 30 μM concentrations. All were synthesized to a chemical purity of >99.5%, as assessed by chemical characterization by a combination of 1H NMR, 13C NMR and GC/MS analyses (Bruker AC-300 spectrometer, together with elemental analyses (Atlantic Microlab, Inc., Norcross, GA), as previously described [42].
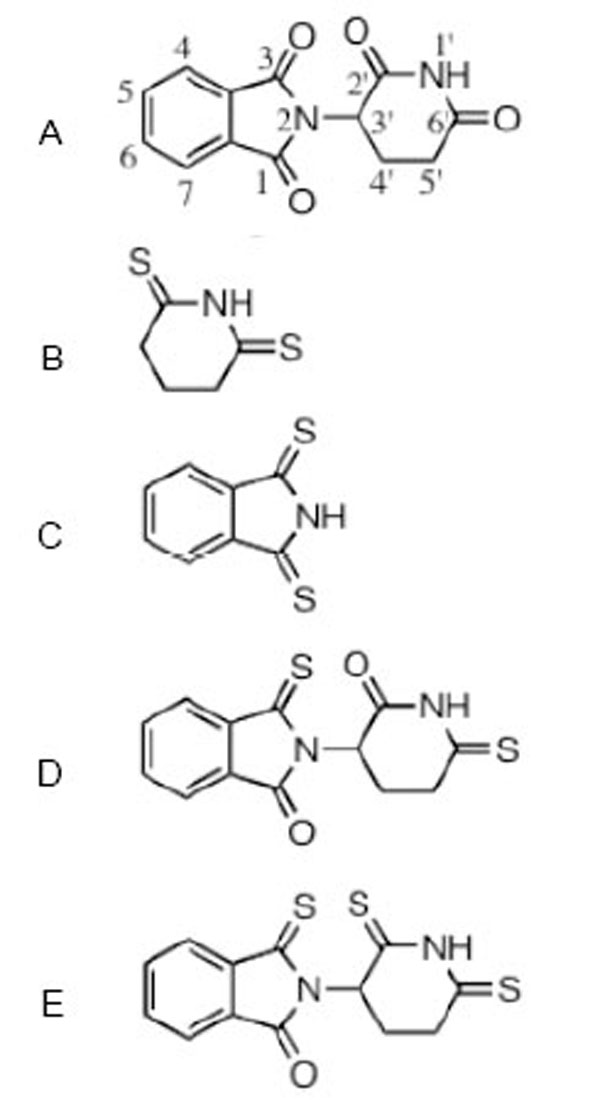
The structure of thalidomide and thio-thalidomide analogs used in this study are presented: A thalidomide (C logD -0.83); B dithioglutarimide (C logD -0.54); C dithiopthalimide (C logD 0.72); D 3,6’-dithiothalidomide (C logD -0.86); E 3,2’, 6’- trithiothalidomide (C logD -0.30). C logD is an index of lipid solubility of an agent, negative values indicate a preference for a lipid environment.
Statistical Analyses
Data throughout are expressed as means ± standard errors, where the n number is shown in parentheses. Statistical comparisons were undertaken by use of either a Students t-test, or by One Way ANOVA with appropriate Bonferroni corrections for multiple comparisons, as required (GraphPad InStat Version 3.05). P values of <0.05 are considered to be of statistical significance, *, **, *** refers to P<0.05, P<0.01 and P<0.001 respectively.
RESULTS
TNF-α and Nitrite Generation
Utilizing optimized RAW 264.7 cell culturing conditions initially designed to assess the effects of novel thalidomide analogues on LPS-induced TNF-α synthesis [43, 44], we attempted to measure the levels of nitrite generated over a 24 hour incubation period, where the cells were challenged with a range of LPS concentrations. This experiment indicated that the administration of LPS, at concentrations up to 60 ng/ml for 24 hours, to RAW 264.7 cells seeded at 100 x103 cells per well, failed to induce a measurable level of nitrite based on the lower limits of the Griess Reagent System. However, under these same conditions a highly significant increase in TNF-α protein was observed (Fig. 2). The TNF-α levels detected in the culture media increased markedly in a LPS dose-dependent manner, TNF-α levels increased from 484±126 pg/ml for 0.1 ng/ml of LPS up to 11,190±405 pg/ml for 60 ng/ml LPS. Control (unchallenged) TNF-α levels were 203±16 pg/ml. There was no evidence of cell toxicity as indicated by the MTS assay; exposing RAW cells to LPS at concentrations from 0.6 ng/ml and higher caused a small but significant increase in cell proliferation (Fig. 2). The Griess Reagent System assay indicated that the levels of nitrite present in this set of conditioned culture media were below 1.5 µM (data not shown).
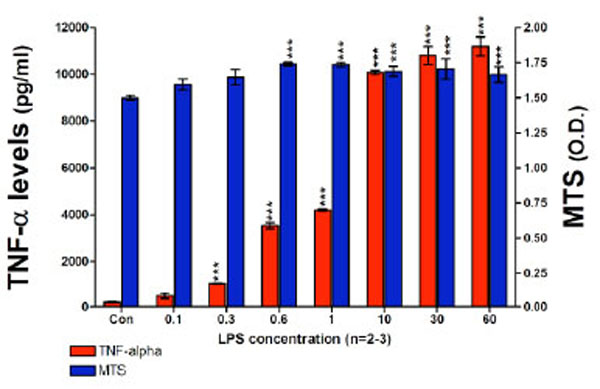
TNF-α protein levels and cell viability after a 24 hour incubation period of RAW 264.7 cells (100K cells/well) with LPS are presented. TNF-α levels (pg/ml) and MTS assay optical density (O.D.) values are shown. TNF-α protein levels detected in the culture media significantly increased in a LPS dose dependent manner from 0.1 ng/ml of LPS to 60 ng/ml of LPS. LPS did not have any adverse effects on cell viability. Data are presented as mean ± S.E.M. of n observations.
In a subsequent set of experiments in order to optimize the cell assay for the measurement of nitrite in culture media we: (1) increased the cell seeding density to 200 x103 cells per well; (2) used higher concentrations of LPS (up to 200 ng/ml of LPS); and (3) employed longer incubation times. After 24 hours incubation with LPS, highly significant elevations in both TNF-α protein and nitrite were measured in culture media (Fig. 3, Top panel). Specifically, TNF-α protein levels ranged from 6913±703 pg/ml for 10 ng/ml LPS, to 15028±1877 pg/ml for 200 ng/ml LPS. By contrast, media from controls unchallenged by LPS demonstrated TNF-α protein levels of 62±6 pg/ml. The nitrite levels ranged from 1.7±0.13 µM for 10 ng/ml LPS to 5.7±0.41 µM for 200 ng/ml LPS. Elevations in nitrite levels appeared to plateau at 130 ng/ml LPS and were similar at LPS concentrations thereafter: 5.7±0.20 µM and 6.4±0.69 µM for 130 and 160 ng/ml, respectively. By contrast, those in control cell media were below the lowest concentration on the standard curve (1.5 µM). The MTS assay indicated no evidence of cell toxicity at the high levels of LPS used in this study. The consequences of a 48 hour incubation of RAW 264.7 cells, 200 x103 cells per well, with LPS on both TNF-α protein and nitrite generation are presented in Fig. (3) (Bottom panel). Specifically, TNF-α levels in the media were elevated to 3348±59 pg/ml for 10 ng/ml LPS, and 7070±128 pg/ml for 200 ng/ml LPS, compared to control levels of 77±4 pg/ml. Correspondingly, nitrite levels ranged from 3.9±0.82 µM for 10 ng/ml LPS to 8.4±0.59 µM for 200 ng/ml of LPS. Nitrite concentrations for 130 and 160 ng/ml of LPS were 9.4±0.77 and 8.8±1.16 µM, respectively. The nitrite levels measured in media from the 48 hours incubation were significantly higher that those measured from the 24 hours time point at corresponding LPS concentrations. Similar to prior studies, nitrite levels for control wells were below the detection limit of the utilized assay and the MTS evaluation indicated a lack of toxicity for all LPS concentrations over 48 hours.
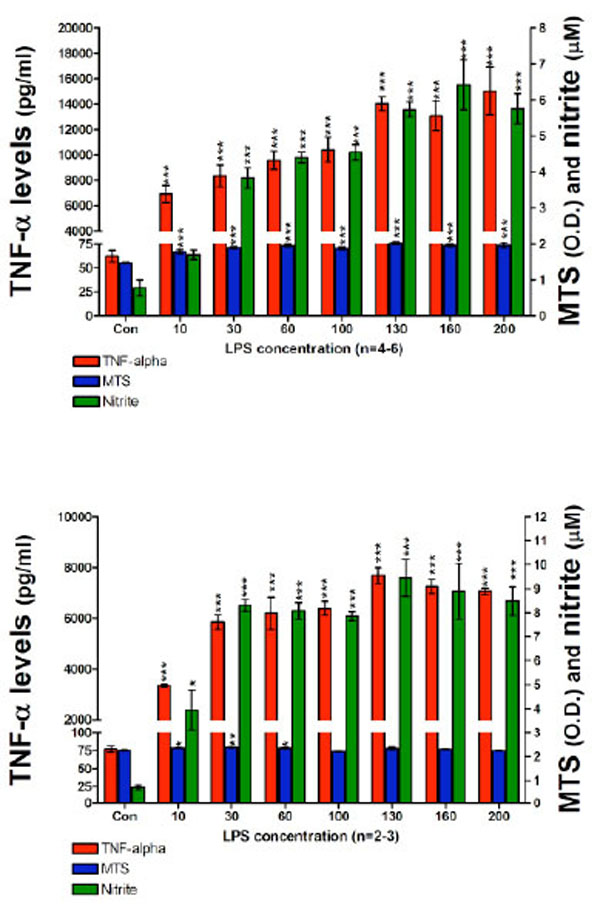
TNF-α protein, nitrite levels and cell viability after a (TOP) 24 hour and (BOTTOM) 48 hour incubation period of RAW 264.7 cells (200K cells/well) with LPS are presented. TNF-α protein levels (pg/ml); MTS assay optical density (O.D.) values and nitrite levels (µM) are shown. After 24 hours (TOP) and 48 hours (BOTTOM) with LPS, 10 ng/ml of LPS to 200 ng/ml of LPS, TNF-α protein levels and nitrite levels measured in harvested media significantly increased in a LPS dose dependent manner. Data from the MTS assay showed that higher doses of LPS displayed no evidence of cell toxicity. Data are presented as mean ± S.E.M. of n observations.
Thalidomide and Analogue Effects on TNF-α and Nitrite Generation
Having characterized the assay conditions, the impact of thalidomide and analogues on LPS induced TNF-α protein and nitrite generation was then examined. At 24 hours following plate seeding with 200 x103 cells per well, pretreatment with thalidomide or analogues (10 or 30 µM) was initiated, and LPS (60 ng/ml) was added an hour later. This LPS concentration was chosen as a submaximal, non-saturating concentration in relation to TNF-α protein and nitrite generation responses (see Fig. 3). A submaximal LPS concentration was chosen to ensure that in addition to drug-induced reductions, elevations in TNF-α protein and nitrite could also be assessed and that both would fall within the linear range of the assay. The following agents were utilized: thalidomide, dithioglutarimide, 3,6’-dithiothalidomide, 3,2’,6’-trithiothalidomide, and dithiopthalidomide, together with drug vehicle (100% DMSO) that was added to control wells. At 24 and 48 hours thereafter, actions on TNF-α protein synthesis, nitrite generation and cell viability were assessed. Evident in Fig. (4) (top panel), assessed at 24 hours thalidomide (10 and 30 µM) had no inhibitory action on secreted TNF-α protein levels when compared to vehicle control cells. Likewise, nitrite levels for thalidomide 10 and 30 µM were no different from vehicle values (control: 2.5±0.05 µM vs. thalidomide 10 and 30 µM: 2.6±0.21 µM and 2.2±0.31 µM), and no changes in cell viability were observed (Fig. 4, Top panel). In contrast, all thalidomide analogues significantly attenuated the LPS-induced rise in TNF-α protein, and similarly reduced generation of nitrite. However, reductions in TNF-α protein and nitrite for specific analogs were associated with a mild loss of cell viability. Dithioglutarimide (10 µM) proved well-tolerated, reduced TNF-α protein levels to 58±14 % of controls and, likewise, lowered nitrite levels to those comparable with control values; from 2.5±0.05 µM to below 1.5 µM (drug values were 0.46±0.10 µM). In the same way, 3,2’,6’-trithiothalidomide (10 µM) lacked toxicity and yet reduced TNF-α protein levels to 48±2 % of control and nitrite to below 1.5 µM, (drug values were 0.03±0.031 µM). The other thalidomide analogues lowered TNF-α protein levels over a range spanning 48±1 % to less than 22±1 % of control values, nitrite values were similarly reduced below the 1.5 µM cutoff; however, the agents were associated with significant, yet variable degrees of cell loss (Fig. 4, Top panel).
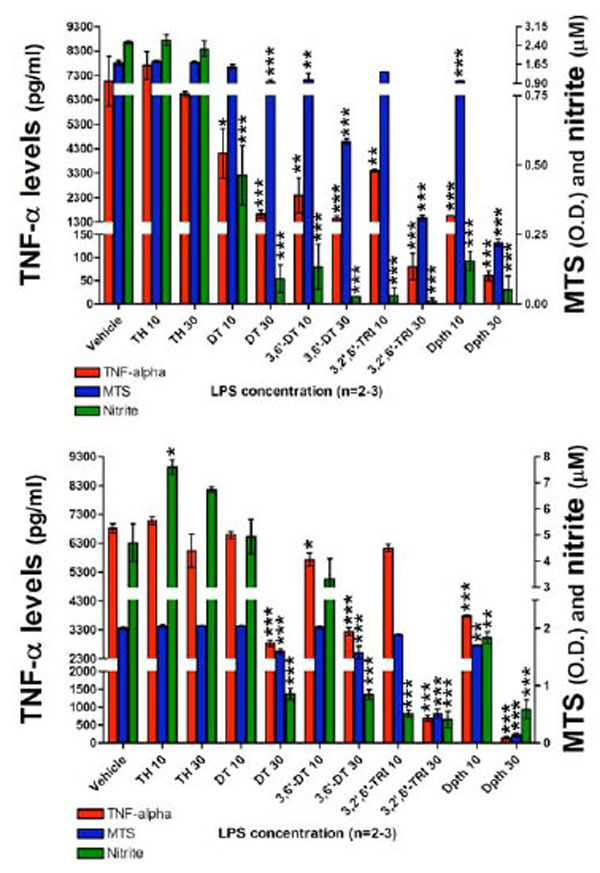
The effects of thalidomide analogs on LPS-induced alterations in TNF-α protein levels, nitrite generation and cell viability of RAW 264.7 cells (200K cells/well) after a 24 (TOP) and 48 hour (BOTTOM) incubation period with LPS are presented. Top panel: 24 hours incubation: TNF-α levels (pg/ml), MTS assay optical density (O.D.) values and nitrite levels (µM) are shown. After 24 hours incubation with drugs and LPS, thalidomide analogs (but not thalidomide) reduced the levels of TNF-α protein. Similarly the generation of nitrite was reduced to below vehicle control values. Both dithioglutarimide and 3,2’,6’- trithiothalidomide at 10 µM, presented anti-TNF-α and nitrite properties with no indication of cell toxicity. The anti-TNF-α and nitrite actions of 3,6’-dithiothalidomide and dithiopthalimide (10 µM) were associated with a degree of cell toxicity. All agents at 30 µM displayed toxicological properties to the RAW cells. Bottom panel: After 48 hours incubation with drugs and LPS, comparable to that evident at 24 hours the thalidomide analogs, but not thalidomide, generated TNF-α and nitrite lowering action. Interestingly, thalidomide at 10 µM significantly elevated generation of nitrite. At 48 hours there was less evidence of cellular toxicity induced by several of the thalidomide analogs. Vehicle refers to 100% DMSO; TH 10 & 30 refers to thalidomide at 10 and 30 µM; DT, 3,6’-DT, 3,2’,6’-TRI and Dpth refer to dithioglutarimide, 3,6’-dithiothalidomide, 3,2’,6’-trithiothalidomide and dithiopthalimide respectively, also at 10 and 30 µM. Data are presented as mean ± S.E.M. of n observations.
Following 48 hours incubation, TNF-α levels in thalidomide (10 and 30 µM) treated cells were no different from vehicle control values (Fig. 4, Bottom panel). Contrary to expectation, thalidomide elevated nitrite generation, raising values from 4.6±0.7 µM (control) to 7.5±0.2 µM and 6.7±0.1 µM at 10 and 30 µM thalidomide, respectively. Similar to that evident at 24 hours, thalidomide analogues proved capable of lowering TNF-α protein and nitrite generation. In this regard, 3,6’-dithiothalidomide and dithiopthalimide at both 10 and 30 µM significantly reduced TNF-α levels. The higher concentration of 30 µM dithioglutarimide and 3, 2’,6’-trithiothalidomide likewise proved able to reduce this cytokine’s level. In relation to LPS-induced nitrite, 10 and 30 µM 3,2’,6’-trithiothalidomide and dithiopthalimide, and 30 µM dithioglutarimide and 3,6’-dithiothalidomide significantly lowered generation. Cell viability data indicated that thalidomide (10 and 30 µM), dithioglutarimide, 3,6’-dithiothalidomide and 3,2’,6’-trithiothalidomide (10 µM) were well tolerated. In contrast, however, the TNF-α protein and nitrite lowering actions of all analogues at 30 µM were associated with a significant reduction in cell viability.
DISCUSSION
The aims of this study were to determine whether novel thiol analogues of thalidomide possessed utility at limiting the synthesis of TNF-α protein and reactive nitrogen species induced by LPS in an in vitro cell model of inflammation. Based on concentration-matched data described here, we extend the prior work of Tweedie et al. [43] and Park et al., [44], and demonstrate that thiol analogues of thalidomide, but not thalidomide, were able to significantly reduce the synthesis of TNF-α protein and consequently the generation of nitrite in a selective and concentration-dependent manner.
The activation of RAW cells by LPS is mediated by the following process: LPS interacts with the CD14/TLR-4/MD2 receptor complex on macrophage / microglial cell surfaces and induces the synthesis of many cellular factors, such as IL-1α/β, IL-6, IL-12 and cyclooxygenase-2 (COX-2), reactive oxygen / nitrogen species (ROS / RNS) and the potent pro-inflammatory protein TNF-α. In the current study, the biological consequences of RAW cell activation by LPS were assessed by measurement of TNF-α protein levels, a marker routinely measured in our laboratory [43], and the levels of a stable end product of nitric oxide metabolism, nitrite (NO2-). TNF-α protein interacts with the TNF-α receptor 1 which regulates NFκB activity, ultimately allowing the cytoplasmic form of the transcription factor to translocate to the nucleus where it can induce a series of target genes involved in inflammation, for a review of the literature see [47, 48]. Classical target genes activated by NFκB are: the cytokines that may well have activated the NFκB pathway (i.e. TNF-α), thus introducing a powerful, cytokine mediated amplification feedback loop; also chemokines and various enzymes such as COX-2, phospholipase A2 (PLA2) and inducible nitric oxide synthase (iNOS or NOS2). The generation of nitric oxide (NO) in macrophage / microglial cells is mediated by the actions of iNOS on L-arginine to form NO and citrulline. However, NO is highly unstable and has a very short half-life of approximately 30 seconds; yet, it can undergo rapid metabolic transformation with reactive oxygen species (i.e. superoxide O2-) to form the highly neurotoxic peroxynitrite, which is associated with cell death [49-51].
As TNF-α signaling is a strong inducer of iNOS–induced NO generation that subsequently allows for the interaction of ROS with RNS intermediates, events that limit the synthesis of TNF-α protein would be predicted to most likely limit the activation of iNOS and reduce the levels of NO metabolites, this was shown to be the case. Thalidomide reduces the half-life of TNF-α mRNA by approximately 50% [38], from additional experiments it was subsequently determined that thalidomide and the thiol analogues reduced the half-life of the mRNA by reducing the RNA stability at the 3’-untranslated region [41, 52]. However, thalidomide and analogues are referred to as immunomodulatory drugs (IMIDs) and clearly impact a host of immune markers, in addition to those described, both by the defined pathways and likely via numerous other primary and secondary ones [44, 53-56].
As shown in these experimental conditions, the levels of measured nitrite are in line with other published reports [44, 57, 58]. Our nitrite values ranged from 1.7 µM up to ~10 µM, depending on the specific experimental condition. It is important to note that the Greiss Reagent System is used to quantify nitrite, and not nitrate, it is highly likely that most of our measurements presented here are underestimations of the true levels of NO intermediate generation. Indeed, Stuehr and Marletta [58] determined that approximately 50 to 75% of the LPS-induced macrophage / microglial cell NO is converted into nitrite and the remainder to nitrate. Therefore, depending on the specific experimental conditions used, our measurements may be limited to as much as a 50% underestimation on total NO generation. Either way, when one considers the implications of these data in light of a chronic setting as seen in most CNS neurological conditions, it is feasible that quantities of nitric oxide generation, as have been reported herein, may be sufficient to induce neurodegeneration.
Thalidomide has been reported to cause apoptosis in cells when used in high concentrations. Such toxicity was not evident at the concentrations utilized in our study. It has additionally been reported that the mechanism underpinning thalidomide-toxicity, as seen in embryonic fibroblasts, is due to the generation of ROS namely; superoxide [59]. Hence, it is likely that thalidomide per se lacks any inherent antioxidant properties. As such, it is feasible to suggest that the anti-nitrite properties of the analogues used in this study were likely not due to any direct antioxidant properties, but due to anti-TNF-α mediated effects. Recent studies have established that thalidomide’s actions on TNF-α are multiple, because it has been described to lower TNF-α under some conditions [38, 60] but not others [61, 62]. In our study, thalidomide at concentrations of 10 and 30 µM did not significantly alter TNF-α, and the lower dose elevated nitrite levels, which in accord with Park et al., [44] appeared to be a more sensitive marker of thalidomide and analog mediated actions. As the thiothalidomide analogs described here can attenuate the levels of both TNF-α protein and the generation of nitrite, a marker of nitric oxide, it is possible they may provide a useful form of treatment in unregulated CNS neuroinflammatory conditions [2, 19, 26, 33] in addition to other disorders [44, 54, 55]. Further evaluation of such agents in models of chronic neuroinflammation will be required to address this question [63].
ACKNOWLEDGEMENT
This work was supported entirely by the Intramural Research Program of the National Institute on Aging, National Institutes of Health. All co-authors report no conflict of interest.
REFERENCES
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link] [PMC Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]

