All published articles of this journal are available on ScienceDirect.

In the Huh7 Hepatoma Cells Diclofenac and Indomethacin Activate Differently the Unfolded Protein Response and Induce ER Stress Apoptosis
Authors Info & Affiliations
Abstract
Non-steroidal anti-inflammatory drugs (NSAIDs) are cyclooxygenases (COXs) inhibitors frequently used in the treatment of acute and chronic inflammation. Side effects of NSAIDs are often due to their ability to induce apoptosis. Located at the Endoplasmic Reticulum membranes a tripartite signalling pathway, collectively known as the Unfolded Protein Response (UPR), decides survival or death of cells exposed to cytotoxic agents. To shed light on the molecular events responsible for the cytotoxicity of NSAIDs, we analysed the ability of diclofenac and indomethacin to activate the UPR in the human hepatoma cell line Huh7. We report that both NSAIDs can induce differently the single arms of the UPR. We show that indomethacin turns on the PERK and, only in part, the ATF6 and IRE1 pathways. Instead, diclofenac reduces the expression of ATF6 and does not stimulate the IRE1 endonuclease, which drives the expression of the prosurvival factor XBP1. Diclofenac, as well as indomethacin, is able to activate efficiently only the PERK pathway of the UPR, which induces the expression of the proapoptotic GADD153/CHOP protein. Our results highlight the importance of the UPR in evaluating the potential of drugs to induce apoptosis.
INTRODUCTION
Non-steroidal anti-inflammatory drugs (NSAIDs) are the most frequently prescribed analgesic and anti-inflammatory agents. The therapeutic efficacy of NSAIDs is due to their ability to impair prostaglandin synthesis by inhibiting cyclooxygenases (COXs) [1]. Despite the beneficial effects, the clinical treatment with NSAIDs frequently produces gastric and duodenal ulcers [2] and citotoxicity in gastric mucosal [3] and liver cells [4, 5] for of their capacity to activate apoptosis [6-10].
Many phases of the caspase cascade induced by NSAIDs, are known, but relatively little has been reported about the upstream events that lead to the activation of the cell death program. Increasing number of evidences show that the Endoplasmic Reticulum (ER) plays a central role in the cell decision between cell survival and death [11]. A signalling pathway, collectively known as the Unfolded Protein Response (UPR), located at the ER membranes replies to cell toxicity induced by protein unfolding within the ER: a condition called ER stress [12-14]. The UPR is coordinated by three transmembrane proteins, negatively regulated by the chaperone BiP/GRP78 [15]: the pancreatic ER kinase (PKR)-like (PERK), the Activating Transcription Factor 6 (ATF6) and the Inositol-Requiring Enzyme 1 (IRE1). Under ER stress, the p90 inactive form of ATF6α (p90 ATF6) dissociates from BiP/GRP78 [16]. This event allows the transport to the Golgi complex of the p90 ATF6α protein, which undergoes to proteolytic cleavage by the Golgi resident site-1 and site-2 proteases [15-17]. Proteolysis at the Golgi complex releases the p50 ATF6α form, which enters the nucleus and activates transcription of target genes [18]. Dissociation from BiP/GRP78 activates the PERK kinase, which phosphorylates the initiation factor eIF2α to attenuate protein synthesis and reduce protein load into the ER [12-14]. In response to ER stress, the endonuclease IRE1 removes 26 nucleotides from the XBP-1 mRNA to unlock the translation of XBP-1, a potent transcription factor that regulates the expression of ER chaperones and factors of the ER associated degradation (ERAD) pathway. As a result of the UPR activation, protein homeostasis is rapidly recovered, but, when the stress is prolonged, or the adaptive response is unsuccessful, the UPR activates the cell death program [19]. IRE1 and PERK can trigger apoptosis with different mechanisms. Independently on its endonuclease activity, IRE1 forms a complex with Traf2 and ASK1 (apoptosis signal regulating kinase-1) that phosphorylates JNK, which, in turn, activates apoptosis [11]. Instead, increased eIF2α phosphorylation driven by PERK favours the expression of the transcription factor ATF4 [20], which, in cooperation with ATF6α, enhances the expression of the C/EBP homologous transcription factor GADD153/CHOP, a pro-apoptotic regulator [21].
To give further insights on the understanding of the molecular events underlying cytotoxic effects we analysed the ability of diclofenac and indomethacin to activate the different arms of the UPR. We show that indomethacin turns on the PERK and, only in part, the ATF6 and IRE1 pathways. Instead diclofenac was able to activate efficiently only the PERK branch of the UPR.
MATERIALS AND METHODS
Antibodies
The antibodies used in immunoblotting are: mouse monoclonal anti-ATF6α (Imgenex Corporation, San Diego CA, USA); rabbit polyclonal anti-GRP78. Cell Signalling Technology, Inc., Danvers, MA, USA); mouse monoclonal anti-CHOP (ABR Affinity Bioreagents, Golden, CO, USA); mouse monoclonal anti-PARP; rabbit polyclonal anti-eIF2α (Cell Signalling Technology, Inc., Danvers, MA, USA); and anti-phospho-eIF2α (Ser51). Cell Signalling Technology, Inc., Danvers, MA, USA; anti-JNK and anti-phosphoJNK (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA); anti-GAPDH mouse monoclonal anti-α-tubulin (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA), anti-rabbit or anti-mouse IgG HRP conjugated secondary antibodies were from Pierce (Thermo Scientific, Rockford, IL, USA).
Cell Culture and Drug Treatment
Human hepatoma Huh7 cells were routinely grown at 37°C in a humidified atmosphere with 5% CO2 in DMEM 10% FCS supplemented with 4.5 g/l D-glucose, 1 mM Na-pyruvate, 2 mM L-glutamine. For the induction of stress, in all the experiments cells were incubated in the presence of diclofenac (Sigma-Aldrich, St. Louis, MO, USA) at a concentration of 300 μM. Indomethacin and thapsigargin (Sigma-Aldrich, St. Louis, MO, USA) were used at a concentration of 500 nM, etopoxide (Sigma-Aldrich, St. Louis, MO, USA) was used at 25 μM.
Western Blot Analysis
Cell extracts were lysed in modified RIPA buffer (Tris-HCl pH 7.4 10 mM, NaCl 150 mM, EDTA 1 mM, NP40 1% Na-deoxycholic 0.1%, PMSF 1 mM, protease inhibitor cocktail). Equal amounts of proteins were separated by 10-12% SDS-PAGE and blotted on ECl Hybond nitro-cellulose membranes (GE Healthcare). Filters were blocked in PBS containing 10% non-fat dry milk and 0.1% Tween-20 and incubated with optimal dilutions of specific antibody for different times depending on the antibodies. Anti-rabbit or anti-mouse IgG HRP conjugated were used as secondary antibody, bands were visualised by autoradiography of ECL reaction (Pierce). Anti α-tubulin or anti-GAPDH antibody were used as control for equal amounts of proteins loaded on the gel.
RNA Extraction and Real Time RT-PCR Analyses
Serial dilutions of total RNA prepared by extraction with Qiazol Lysis Reagent (Qiagen, Germany), were reverse transcribed by AMV Reverse Transcriptase kit (Promega Corporation, Madison, WI, USA) and subjected to Real Time RT-PCR. PCR amplification protocols were performed according to the different sets of primers and analyzed by the semi quantitative method [22]. For PCR amplification the following primers were designed to amplify the corresponding transcripts in human cells: Grp78/Bip-forward: CTG GGT ACA TTT GAT CTG ACT GG; Grp78/Bip-reverse: GCA TCC TGG TGG CTT TCC AGC CAT TC; GAPDH-forward: GAA GGT GAA GGT CGG AGT C; GAPDH-reverse: GAA GAT GGT GAT GGG ATT TC
RT-PCR Analysis of XBP1 mRNA Splicing
RNA was harvested and extracted as above immediately from untreated cells or after the stress treatments. For each sample, 0.5 µg of total RNA were subjected to RT-PCR analysis using the AMV Reverse Transcriptase kit (Promega Corporation). To amplify XBP1 mRNA, PCR was carried out for 30 cycles (94 °C for 2 min; 55 °C for 30 s; and 72 °C for 2 min followed by 10 min at 72 °C) using the following primers: 5’-CCT TGT GGT TGA GAA CCA CC-3’ and 5’-CTA GAG GCT TGG TGTA TA-3’. The 452 and 426 bps fragments representing un-spliced and spliced XBP1 mRNA respectively were detected by staining 4% agarose gels with ethidium bromide.
Flow Cytofluorimetry Analysis
Apoptosis was analysed by propidium iodide incorporation in permeabilized cells and flow cytometry [23]. Cells (5 x 104) were cultured in 24-wells plates. After 24 h, drugs were added at different doses and cells were re-cultured for different times (24, 48 and 72 h). For apoptosis analysis, permeabilized Huh7 cells were labelled with propidium iodide (PI) by incubation at 4°C for 30 min with a solution containing 0.1% sodium citrate, 0.1% Triton X-100 and 50 µg/ml PI (Sigma-Aldrich, St. Louis, MO). The cells were subsequently analysed by flow cytometry by a FACSCalibur flow cytometer (Becton Dickinson, North Ryde, NSW, Australia). Each determination was repeated three times.
RESULTS
Diclofenac and Indomethacin Induce Low Level of Grp78 mRNA Expression
Grp78 is a reliable indicator of the ER stress and its induction is clear evidence that the UPR is being activated. Thus, to examine the potential of diclofenac (DIC) and indomethacin (IND) to induce the UPR in the Huh7 cells, we measured the level of mRNA accumulation encoding the molecular chaperone Grp78 (Fig. 1). To that purpose, we performed semi quantitative RT-PCR analyses [22] on RNA samples obtained from untreated Huh7 cells or from the same cells treated with DIC or IND or thapsigargin (TG). In all the experiments, the GAPDH mRNA was used as reference gene for constitutively expressed mRNA. Results of RT-PCR analyses showed that DIC treatment induced a weak accumulation of the PCR fragment amplified from the Grp78 mRNA, with a maximal level after 8 h of exposure (Fig. 1). In addition, level of DIC-induced Grp78 was lower then the one shown by the IND-induced cells. On the other hand, the cells expressed higher level of Grp78 mRNA in response to TG, a powerful ER stress inducer that is able to activate all three sensors of the UPR [24].
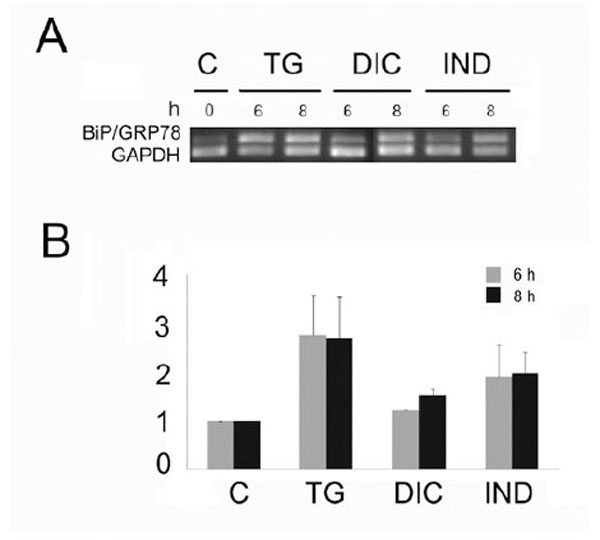
Level of the Grp78 mRNA in Huh7 cells exposed to diclofenac (DIC), indometacin (IND) and thapsigargin (TG). A: Total RNA fractions obtained from cells exposed for the times indicated in hours (h) to 300 µM diclofenac (DIC), 500 nM indomethacin (IND) or thapsigargin (TG) were analyzed by semi quantitative RT-PCR to measure the level of the indicated mRNAs. The panel shows the inverted photographs of DNA fragments stained with ethidium bromide.
B: Histograms represent values of densitometry analysis obtained by the Image J program. The relative fold of induction was calculated as the ratio of treated cells divided by untreated cells. Relative folds of accumulation were obtained by normalising the signals for GAPDH mRNA expression. Each value represents the mean±SE of three independent experiments.
Different Involvement of the Transcription Pathways of the UPR ATF6 and XBP1 in Response to Diclofenac and Indomethacin
Since Grp78 is under the transcriptional control of the UPR, the results obtained by measuring of the rate of Grp78 mRNA accumulation in the DIC- and IND-induced cells, prompted us to investigate the ability of both compounds to activate the different branches of the UPR. To that purpose we analysed by immunoblotting the rate of p90 ATF6α processing into the cleaved p50 ATF6α in the cells induced by DIC or IND (Fig. 2A). In these experiments, the rate of DIC- or IND-induced p50 ATF6α was compared to that measured in the TG-induced and in control cells. Our results showed that in the untreated cells, barely detectable level of the p50 ATF6α form was revealed (Fig. 2A), while, with different intensities, the cleaved form was detectable in both DIC- and IND-treated cells. Instead, in the DIC-induced cells we observed decreased amount of the p90 ATF6α protein and, as a consequence, reduced level of the p50 cleaved form, suggesting that expression of ATF6α is impaired in DIC-induced cells.
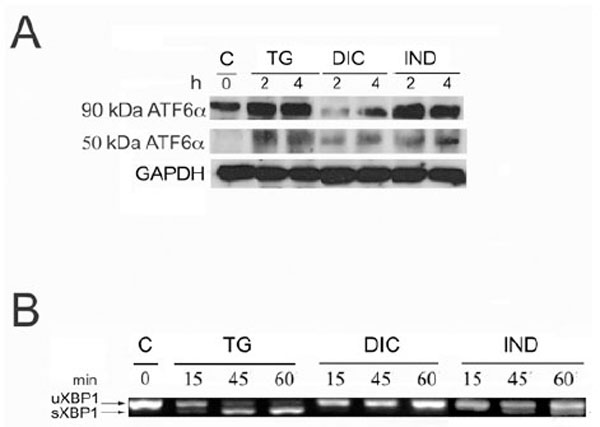
Activation of ATF6α and XBP1 in response to diclofenac and indometacin. A: Protein extracts from control, thapsigargin- (TG), diclofenac- (DIC) and indomethacine- (IND) treated Huh7 cells for the times indicated in hours (h) were analyzed by SDS-PAGE and Western blot. To detect the low level of endogenous ATF6α, 50 mg of protein extract were loaded on the SDS-PAGE. Arrows indicate uncleaved 90 kDa- and cleaved 50 kDa-ATF6α. B: Total RNA samples extracted from Huh7 cells exposed for the times indicated in minutes (min) to either diclofenac (DIC) or indomethacin (IND) were analyzed by RT-PCR to detect the XBP1 mRNA splicing forms. The migration on the gel of the 452 bps un-spliced fragment of the XBP1 mRNA (XBP1 u) and of the spliced 426 bp fragment (XBP1 s) is indicated.
To further investigate the effect of DIC and IND on the transcription pathways of the UPR, we analyzed the level of IRE1 endonuclease activity in the DIC-, IND- and TG-treated cells (Fig. 2B). The activity of IRE1 was measured by comparing the amount of the 426 nts PCR fragment amplified by RT-PCR from the active spliced form of XBP1 mRNA in total RNA samples (Fig. 2B). The results of the RT-PCR analysis showed the appearance of the 426 PCR fragment amplified from the spliced XBP1 mRNA following 15 min of TG treatment with a maximal level of accumulation at 60 min, proving that the mRNA processing in response to TG is very rapid and effective (Fig. 3). In contrast, in the DIC-induced cells the RT PCR analyses revealed only the un-spliced 452 form (Fig. 2B: XBP1-u), suggesting that the IRE1 endonuclease is not activated in the DIC-treated cells. Finally, the spliced form of the XBP1 mRNA (Fig. 2B: XBP1-s) was moderately expressed in the IND-treated cells suggesting that, although to small extent, indomethacin induces the IRE1 endonuclease activity.
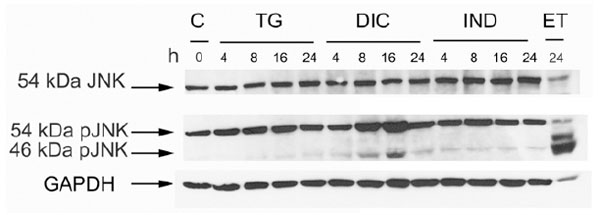
Phosphorylation level of the JNK protein in the response to thapsigargin (TG) diclofenac (DIC) and indomethacin (IND) in Huh7 cells. Western blot analysis of cell extracts obtained from Huh7 cells exposed to TG, DIC or to IND for the times indicated. Etopoxide was used as positive control for the JNK protein phosphorylation. The blots were probed with antibodies against the unphosphorylated (JNK) or phosphorylated (pJNK) JNK proteins.
Diclofenac Induces the Phosphorylation of Thec-Jun N-Terminal Kinase (JNK)
In a mechanism that is independent from its endonuclease activity IRE1 promotes apoptosis, by activating the c-Jun N-terminal Kinase (JNK) signalling pathway [25]. Therefore, we analysed by western blotting the level of phosphorylated JNK accumulating in the cell extracts obtained from untreated Huh7 cells or from the same cells exposed to DIC, or IND or TG. Immunoblots were revealed by anti-JNK antibody (JNK), which detects the un-phosphorylated isoform (54 KDa JNK) of the protein, and by p-JNK antibody, which detects the phosphorylated 54 KDa and 46 KDa isoforms of JNK. In all the experiments, GAPDH was used as a control for constitutively expressed protein (Fig. 3). The results showed that either TG- or IND-treatment induced barely detectable level of JNK phosphorylation. Instead, DIC was able to stimulate higher level of phosphorylation suggesting that diclofenac is an efficient inducer of the signalling pathways that activate JNK.
Diclofenac as well as Indomethacin Activate the PERK Pathway of the UPR
To analyse PERK activity, we compared by western blotting the level of phosphorylated eIF2α accumulating in the cell extracts obtained from untreated Huh7 cells or from the same cells exposed to DIC, or IND or TG. Immunoblots were revealed by the eIF2α antibody (eIF2α) to detect the un-phosphorylated form of the factor, and the eIF2α-P antibody to detect the phosphorylated form of the protein. In all the experiments, α-tubulin was used as a control for constitutively expressed proteins. The results, shown in Fig. (4A), revealed that either DIC or IND were able with similar time and intensity to induce eIF2α phosphorylation, suggesting that DIC as well as IND are powerful inducers of the PERK kinase activity. In addition, our results showed that DIC- and IND-activation of PERK is very fast (15-30 min), progressively reached the maximal level at 120 min of drug treatment and remained stable for up to 6 hours (data not shown).

Activation of the PERK pathway in response to diclofenac and indomethacin. A: Western blot analysis of protein extracts obtained as above and revealed by eIF2α antibody. Immunoblot were revealed by antibodies recognising un-phosphorylated form of the eIF2α protein (eIF2α) or the phosphorylated form of the eIF2α protein (P-eIF2α) and tubulin (TUB). B: Accumulation of the CHOP protein in response to thapsigargin (TG) diclofenac (DIC) and indomethacin (IND) in Huh7 cells. Immunoblots of cell extracts obtained from Huh7 cells induced with TG, DIC or to IND for the times indicated. The blots were probed with antibodies against CHOP or tubulin (TUB) as a control of equal loading of proteins on the gel.
Increased eIF2α phoshorylation induces the ATF4/CHOP pathway of the ER stress apoptosis. Thus, we examined the expression of the GADD153/CHOP protein in the DIC-, IND- and TG-induced cells. Results of the immunoblots showed that the GADD153/CHOP protein (CHOP) was absent in the untreated cells (Fig. 4B). As we would expect, CHOP accumulated in the DIC-, IND- and TG-induced cells within 8 h of drug exposure.
Diclofenac and Indomethacin Induced Similar Level of Apoptosis in the Huh7 Cells
To measure the level of DIC- and IND-induced apoptosis we performed flow cytometric analysis in Huh7 cells treated for different times. Results showed the percentage of the apoptotic nuclei increased in a time-dependent manner after drug treatment of the cells (Fig. 5). Interestingly, the lowest rates of apoptotic nuclei were observed in the cells exposed to etopoxide, which induces cell death with mechanisms that are distinct from the ER stress. In contrast, for the TG-, DIC- and IND-induced cells we observed a higher rate of cell death, which occurred between 48 and 78 h of each drug treatment, suggesting that diclofenac and indometacine, as well as thapsigargin can induce cell death at similar rates.
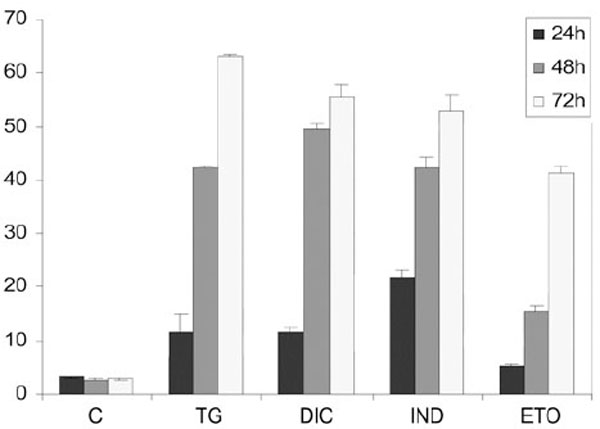
Flow cytometry analysis of the rates of apoptosis in Huh7 cells exposed to DIC or to IND. For apoptosis analysis, permeabilized Huh7 cells were labelled with propidium iodide (PI) and analysed by flow cytometry by a FACSCalibur flow cytometer (Becton Dickinson, North Ryde, NSW, Australia). DNA damaging agent etopoxide was used as positive control for apoptosis. Results are the mean of three experiments repeated three times.
DISCUSSION
In this study we describe how the different arms of the UPR might contribute in the cytotoxicity of indomethacin and diclofenac, two widely used NSAIDs.
First, the evidences here reported show that diclofenac and, at lesser extent indomethacin, are able to induce only partially the expression of the Grp78 mRNA. This observation suggests that both compounds do not fully activate the transcriptional pathways of the UPR. In particular, in the diclofenac induced cells, we observed decreased level of either the p90 and the p50 ATF6α cleaved form, which suggests that the expression of ATF6, is at some extent, impaired by diclofenac.
Grp78 is the master regulator of the UPR, which is controlled by the chaperone through the binding-release of the ER stress transducers IRE1, PERK and ATF6α. Moreover, Grp78 retains anti-apoptotic properties for its ability to interfere with caspase activation [26, 27]. Thus, GRP78 induction under ER stress represent a major cellular protective response for the cell to survive. Therefore, the little level of Grp78 observed in the DIC- and, at higher extent, in the IND-induced cells, could be inadequate to activate the survival mechanisms required to counteract ER stress induced by diclofenac.
Interestingly, our results show the IRE1/XBP1 pathway of the UPR is blocked in the diclofenac treated cells. Consequently in these cells the transcription factor XBP1 is not expressed. XBP-1 is a potent activator that regulates the expression of genes of the ER associated degradation (ERAD) pathway [28], such as the ER degradation-enhancing mannosidase-like protein (EDEM) [29]. Moreover, retains prosurvival properties. Overexpression of XBP-1 allows cell survival in solid tumors, in which cells grow under severe hypoxia [30], and ita has been described that in XBP-1 deficient cells increased apoptosis is observed under stress conditions [31]. Thus, in the absence or reduced IRE1 endonuclease activity, as observed during cell exposure to diclofenac and indomethacin, cell survival can be severely reduced because of the lack of XBP1 expression.
IRE1 mediates two responses, which independently may give opposite contributions in the cell decisions between life and death: the splicing of XBP1 mRNA and the activation of the c-JUN N-terminal Kinases (JNK) [32]. In this context, a number of evidences show that the splicing of XBP1 is down regulated when apoptosis is induced, and that the overexpression of XBP1 during the late stages of ER stress induced apoptosis, protects cells from death [33]. On the basis of the above considerations, it seems very likely that in the case of diclofenac the lack of IRE1 endonuclease activity could favour the IRE1/JNK apoptotic pathway giving rise to the higher level of JNK phosphorylation observed in the DIC-induced cells.
Otherwise, the c-JUN N-terminal Kinases (JNK) are activated by various stimuli, including UV light, interleukin-1, tumor necrosis factor-α (TNF-α). Therefore, we cannot exclude that the higher level of phosphorylated forms of JNK could be the result of apoptotic signalling pathways activated by diclofenac independently on the ER stress.
Our results show that diclofenac, as well as indomethacin, activates the PERK pathway of the UPR. This early event has a primary cell defensive role. Phosphorylation of eIF2α by PERK reduces general translation and, therefore, the load of newly synthesized proteins, which allows the ER to better manage stress and successfully recover protein unfolding. However, this response appears to be only a timely restricted protection. Prolonged activation of the PERK/eIF2α pathway can induce the switch from cell survival to apoptosis by activating the expression of the GADD153/CHOP proapoptotic protein. GADD153/CHOP is a transcription factor that down regulates the expression of Bcl-2 and up regulates the expression of other proapoptotic members of the Bcl-2 family [34, 35]. By our experiments, we found that either diclofenac or indometacin induced high level of the apoptotic executor GADD153/CHOP. Increased expression of GADD153/CHOP has been reported to lead to cell cycle arrest and apoptosis [36] and our results confirm that the ER stress induced protein GADD153/CHOP is involved in NSAID-induced apoptosis [37].
In conclusion, our work provides a comprehensive picture of the contribution of each arm of the UPR on the cell toxicity induced by NSAIDs. The UPR response could be a helpful instrument to predict the potential of drugs to induce cell toxicity and could be useful to select non-toxic derivatives. However, our results suggest that much remains unknown, as, for example, how is UPR regulated to switch from a protective response to a pro-apoptotic response leading to cell death.
ACKNOWLEDGEMENTS
This work was partly supported by FARB grants 2008 and 2009 from the University of Salerno to P. R.
REFERENCES
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]

