All published articles of this journal are available on ScienceDirect.

Conformational States and Kinetics of the Calcium Binding Domain of NADPH Oxidase 5
Authors Info & Affiliations
Abstract
Superoxide generated by human NADPH oxidase 5 (NOX5) is of growing importance for various physiological and pathological processes. The activity of NOX5 appears to be regulated by a self-contained Ca2+ binding domain (CaBD). Recently Bánfi et al. suggest that the conformational change of CaBD upon Ca2+ binding is essential for domain-domain interaction and superoxide production. The authors studied its structural change using intrinsic Trp fluorescence and hydrophobic dye binding; however, their conformational study was not thorough and the kinetics of metal binding was not demonstrated. Here we generated the recombinant CaBD and an E99Q/E143Q mutant to characterize them using fluorescence spectroscopy. Ca2+ binding to CaBD induces a conformational change that exposes hydrophobic patches and increases the quenching accessibilities of its Trp residues and AEDANS at Cys107. The circular dichroism spectra indicated no significant changes in the secondary structures of CaBD upon metal binding. Stopped-flow spectrometry revealed a fast Ca2+ dissociation from the N-terminal half, followed by a slow Ca2+ dissociation from the C-terminal half. Combined with a chemical stability study, we concluded that the C-terminal half of CaBD has a higher Ca2+ binding affinity, a higher chemical stability, and a slow Ca2+ dissociation. The Mg2+-bound CaBD was also investigated and the results indicate that its structure is similar to the apo form. The rate of Mg2+ dissociation was close to that of Ca2+ dissociation. Our data suggest that the N- and C-terminal halves of CaBD are not completely structurally independent.
INTRODUCTION
Reactive oxygen species (ROS) such as nitric oxide, superoxide, and hydrogen peroxide have received much attention recently due to their implications in physiology and pathophysiology [1]. Superoxide is primarily generated by the enzyme family of NADPH oxidases (NOXs), and there are several homologous enzymes identified so far, including NOX1, NOX2, NOX3, NOX4, NOX5, and dual oxidase [2, 3]. Among them, NOX2 is well known for its pathogen-killing ability in phagocytes, in which a large amount of superoxide is generated through the NOX2 multienzymatic system [4]. While such a large amount of superoxide is essential in phagocytic cells, superoxide generated from other non-phagocytic cells has been implicated in intracellular signaling leading to altered gene expression and protein biosynthesis, apoptosis, migration, hypertrophy, kinase activation, oxygen sensing, bone resorption, and extracellular matrix modification [5]. Accordingly, deficiency or overproduction of superoxide has been demonstrated to lead to pathological problems, as well as neurological, cardiovascular, or renal diseases [6-8].
The exact biological functions and superoxide-generating mechanisms of non-phagocytic NOXs are still unclear. Like the NOX2 system, NOX1 and NOX3 require accessory proteins such as NOXA1, p47phox, and/or p67phox to form a multi-enzyme complex for superoxide production [2]. Interestingly, the NOX5 activity is regulated by Ca2+ flux, which was demonstrated in vivo [9] and in vitro [10]. NOX5 was detected in all fatal tissue, and is expressed in adult spleens, testis, ovary, placenta, pancreas [3, 9], sperm, and while blood cells [11]. NOX5 consists of a gp91phox homologous flavoprotein domain that binds FAD and NADPH, a heme-binding transmembrane domain, and a Ca2+-binding domain (CaBD). Based on the model of NOX2 [12], the hypothesized mechanism for NOX5 is that Ca2+ binding causes a conformational change on CaBD, exposing hydrophobic patches, and leading to interactions between the CaBD and flavoprotein domains, which allows for electron transfer from NADPH to molecular oxygen. The physical interaction between the CaBD and flavoprotein domains has been demonstrated by pull-down method [10]. Based on this model, NOX5 contains all essential components for superoxide production. However, the high concentration of Ca2+, approximately 40 μM, needed to saturate CaBD and activate NOX5 superoxide production in vitro makes such Ca2+-activation mechanism nonexclusive because it is unclear whether such concentration can be met in stimulated cells. Indeed, recent studies indicate that the NOX5’s calcium sensitivity is enhanced by the interaction with calmodulin (CaM) [13] and by a phosphorylation mechanism [14]. Information on how Ca2+ and other factors influence the electron transfer within NOX5 is essential for understanding its superoxide-producing mechanism.
CaBD contains four Ca2+ binding sites, forming two tandem pairs with one non-canonical and three canonical EF-hand motifs. Its Ca2+ binding has been investigated by flow dialysis and the results indicate that its N-terminal half containing the 1st and 2nd EF-hand motifs has a low Ca2+ binding affinity (Kd = 15-20 μM), while the C-terminal half containing the 3rd and 4th EF-hand motifs has a high Ca2+ binding affinity (Kd = 3.75-3.80 μM) [10]. The conformational change of CaBD induced by Ca2+ binding appears to be very important for it to interact with the NOX5’s flavoprotein domain. Previously, this conformational change was monitored by intrinsic tryptophan (Trp) fluorescence and 2,6-p-toluidino-naphthalene sulfonate (TNS) binding for hydrophobic content [10]. However, neither an examination of the conformational change of CaBD upon Mg2+ binding, nor an investigation of kinetics about metal ion association and dissociation for CaBD has been reported. Here we utilized techniques of fluorescence quenching, 8-anilino-1-naphthalene sulfonic acid (ANS) hydrophobic binding, and chemical-induced unfolding monitored by intrinsic and extrinsic fluorescence to characterize CaBD. Furthermore, the secondary structure of CaBD was determined by CD spectroscopy, and the kinetics of metal dissociation from CaBD was determined using stopped-flow spectroscopy.
MATERIALS AND METHODS
Reagents
All chemicals were purchased from Sigma-Aldrich (St. Louis, MO) and Fisher Scientific (Pittsburgh, PA), and used without further purification. All buffers used in the protein purification and the subsequent spectroscopic characterization were rendered “Ca2+-free” by treatment with Chelex-100 (Bio-Rad Laboratories, Hercules, CA). The spectroscopic measurements were performed at least three times using at least three different protein batches and were reproducible.
Plasmid Constructions
The pLW vector, a generous gift from Dr. Lucy Waskell at the University of Michigan, was digested with Nco I and Xho I, and a phosphorylated double stranded oligonucleotide (5’-CATGGCGCATCATCACCATCACCATGCGGTAC and 5’- CGCATG GTGATGGTGATGATGCGC) that encodes peptide sequence MAHHHHHAV and contains Nco I and Kpn I compatible sites (underlined), flanking 5’ and 3’ ends, was ligated to the digested pLW vector to create a linear pLW-His6 vector containing Kpn I and Xho I cloning sites. The cDNA encoding human NOX5β was a generous gift from Dr. Botond Bánfi at the University of Iowa. The CaBD domain (residues 1-184) was amplified by polymerase chain reaction (PCR) with the primers: 5’-GACTGGTACCG ATGAGTGCCGAGGAG) and 5’-CGCACTCGAGTTAGG CGCGGGTCAGCTG. The forward and reverse primers introduced two restriction sites (underlined) for Kpn I and Xho I sites on both ends and a stop codon in the 3’ end. The PCR product was subcloned into the above pLW-His6 vector to create the plasmid pLW-His6CaBD. The recombinant protein was constructed in such way that it contains an additional eleven amino acids, MAHHHHHHAVP, on its N terminus. The plasmid containing the mutant E99Q/E143Q was generated using a QuickChange Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA). Two sequential site-directed mutations were performed by first generating the single mutation E99Q, and then E99Q/E143Q. The clones were sequenced to confirm the mutations and the oligonucleotide primers were synthesized by Integrated DNA Technologies (Coralville, IA).
Recombinant Protein Expression and Purification
The plasmids pLW-His6CaBD or pLW-His6E99Q/E143Q were introduced into BL21(DE3) E. coli cells. Cells were grown in Luria−Bertani (LB) medium to OD600 0.5-0.7, and the expression was induced by the addition of 1 mM isopropyl-β-D-thiogalactopyranoside (IPTG) at 37 oC for 4 h. All of the following purification steps were performed at 4 oC. The histidine-tagged recombinant proteins were first purified with a Ni2+-chelating Probond column (Invitrogen Carlsbad, CA). Briefly, after harvest cells were resuspended in a buffer containing 50 mM Tris, pH 7.5, 0.5 M NaCl, 10 mM Imidazole (Buffer A) followed by homogenization and centrifugation. The resulting supernatant was loaded on a Probond column equilibrated with Buffer A. The column was washed with the same buffer until OD280 reached baseline. The target protein was eluted with Buffer A containing 300 mM imidazole. Protein was further purified with a phenyl-sepharose column (GE Healthcare BioSciences, Piscataway, NJ) as previously reported for CaM [15]. The purified proteins were then buffer-exchanged in an Amicon Ultra centrifugal filter device (5,000 Da MWCO, Millipore, Bellerica, MA) with a buffer containing 50 mM Tris, 0.125 M NaCl, 5 % glycerol, 1 μM EDTA, pH 7.5 and stored at -80 oC until use. The protein purity was estimated > 95 % based on density profiles measured using UN-SCAN-IT software (Silk Scientific, Inc, Orem, UT).
Preparation of AEDANS-Labeled CaBD and E99Q/E143Q
Approximately 100 μM purified CaBD or E99Q/E143Q in 50 mM EPPS, pH 7.5 was incubated with 2 mM 5-((((2-iodoacetyl)amino)ethyl)amino)naphthalene-1-sulfonic acid (IAEDANS, Invitrogen) in the presence of 1 mM Ca2+ at 4 oC overnight in the dark. Iodoacetamide was added to the reaction mixture (final concentration was 1 mM) to quench the reaction. The free dye and quencher molecules were removed by passing the mixture through a Ni2+-chelating column, and the AEDANS-labeled protein was recovered by eluting with a 0.3 M imidazole solution. The stoichiometry of the labeling was determined to be 0.93 ± 0.11 based on the protein assay kit (Bio-Rad Laboratories) for proteins and the absorption coefficient (5,700 cm-1M-1) for IAEDANS.
Spectroscopic Measurements
Absorption measurements were carried out using a Cary-3 UV/vis double-beam spectrometer (Varian, Inc., Lake Forest, CA). The fluorescence spectra were recorded on a FluoroMax-3P (Horiba John Yvon, Inc., Edison, NJ) equipped with excitation/emission polarizers and a temperature control unit. In all cases, the fluorescence spectra were typically obtained using 2-10 μM protein with the excitation wavelengths at 295, 340, and 350 nm for Trp, AEDANS, and ANS fluorescence, respectively, at 20 oC. The window widths of 2 and 5 nm for the entrance and exit slits were chosen to eliminate photobleaching. All reported spectra were corrected for buffer effects, dilution factors, and any wavelength-dependent response for the fluorometer.
Stern-Volmer Quenching
Intrinsic and extrinsic fluorescence quenching experiments were carried out at 20 oC by adding a stock solution of 6 M acrylamide, which was determined using the absorbance coefficient ε295=2.5 M-1 cm-1, to the sample solution containing 4-10 μM protein and 50 mM Tris, 0.125 M NaCl, pH 7.5. The following Stern-Volmer quenching equation was used for fitting
where F and Fo are the fluorescence intensities at a given concentration of quencher and in the absence of quencher, respectively, Ksv is the dynamic or collisional quenching constant, and [Q] is the concentration of added quencher.Far-UV Circular Dichroism (CD)
The CD spectra were recorded using a JASCO J-715 instrument (JASCO Corporation, Japan) equipped with a temperature control unit. CD spectra were recorded using a cylindrical 0.1 cm path quartz curve and were shown as the average of 3 scans using a spectra bandwidth of 1.0 nm. In all measurements, 0.1 mg/ml proteins in 10 mM Tris, pH 7.5 with 1 mM Ca2+, 10 mM Mg2+, or 0.5 mM EDTA were scanned from 190 nm to 260 nm.
Kinetics of Metal Dissociation
The kinetics was measured using the FluroMax-3P with a stopped-flow accessory SFA-20 (Tgk Scientific, Bradford-on-Avon, UK). To determine the calcium dissociation rate constant, a solution containing 5 to 10 μM unlabeled protein (or 2 to 5 μM labeled protein) and 100 μM Ca2+ (or 2 mM Mg2+ plus 1 mM EGTA) was rapid-mixed with a solution containing EDTA ranging from 10 to 20 mM EDTA at 20 oC. Ca2+ or Mg2+ dissociation was monitored by fluorescence decay. Six to eight injections were averaged to generate the final traces shown in Fig. (5). The integration time and time interval were 2 and 5 ms, respectively. Since the data acquisition for our instrument is triggered after the stopped syringe meets the stop block, the points collected in the original data set are tens of ms later after mixing. The following single-exponential decay equation was used to fit the data
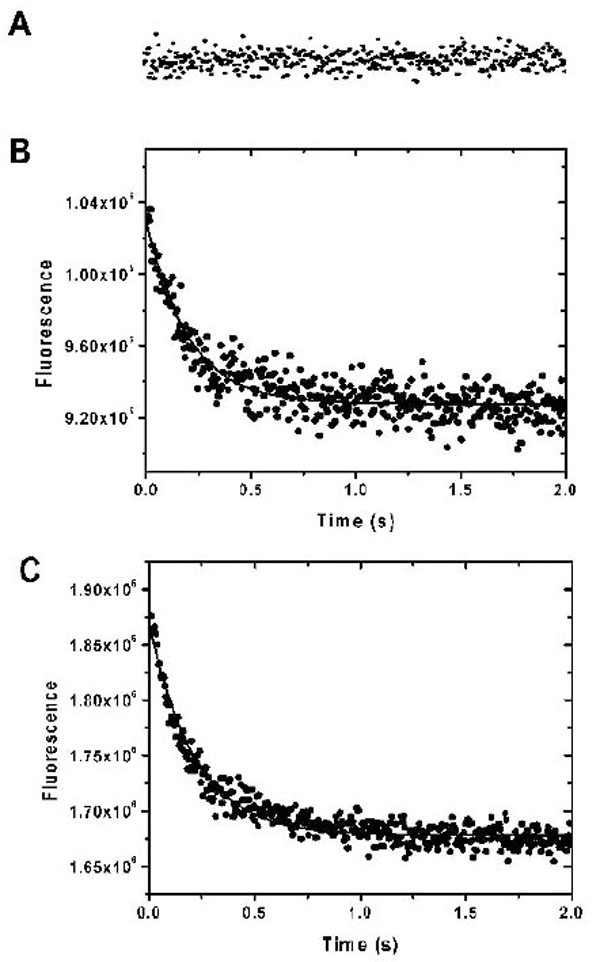
Ca2+-bound protein was quick-mixed with a solution containing large excess of EDTA and its fluorescent decay at 460 nm was recorded with the excitation wavelengths of 340 nm. (A) Shows the kinetic trace of Ca2+ dissociation from AEDANS-E99Q/E143Q, where (B) and (C) show the kinetics of Ca2+ and Mg2+ dissociation from AEDANS-CaBD. The traces are representative of four to six independent experiments each.
Chemical Stabilities
To determine chemical stabilities, approximately 10 μM unlabeled or 5 μM AEDANS-labeled protein was incubated in a buffer containing 50 mM EPPS, pH 8.0, 500 μM EDTA (1 mM Ca2+ or 10 mM Mg2+), and guanidine hydrochloride (Gdn) ranging from 0 to 6 M for at least 6 h. The samples were excited at 295 nm (340 nm for the labeled proteins), and the wavelengths of their emission were recorded. The maximum emission wavelengths of AEDANS-CaBD and AEDANS-E99Q/E143Q induced by Gdn were fitted into eq.3 to determine the fraction of unfolded protein (fU), assuming a two-state unfolding model
where FN, FD, and Fobs represent the maximum emission wavelengths in the native state, unfold state, and in the presence of a given concentration of the denaturant ([D]), respectively. Because F is linearly dependent on the Gdn concentration in the native and unfolded states, FN and FU were determined from the baselines prior and after the unfolding transition using where mN and mD are the slopes, and YN and YD are intercepts.For chemical stability determined by Trp fluorescence, the fu was simply calculated as
where the λobs is the maximum wavelength of the protein at specific concentration of Gdn, λ0M and λ6M are the maximum wavelengths of the protein in the absence of Gdn and in the presence of 6 M Gdn, representing the completely folded and unfolded states.RESULTS AND DISCUSSION
Effects of Metal Ions on the Fluorescence Spectra of CaBD
Previously Bánfi et al. had expressed the recombinant CaBD (residues 1-169) of NOX5 in E. coli, in which it was initially expressed as a glutathione S-transferase (GST) fusion protein followed by thrombin cleavage [10]. Since CaBD contains three tryptophan (W9, W12, and W165) and one tyrosine (Y87), it is suitable for a study using fluorometry. It had been demonstrated that its Trp fluorescence intensity was enhanced upon Ca2+ binding. Interestingly, the fluorescence of its half domains, termed as N-CaBD (residues 1-78) and C-CaBD (residues 79-169), did not respond Ca2+, and therefore it was concluded that the resulting fluorescence enhancement was only associated with the relative conformational change of the two half domains. To eliminate the cleavage step used in the previous study that limits the amount of purified protein for structural and functional studies, we placed a six-histidine tag at the N-terminus of CaBD and purified it to be homogeneous. Our approach generated a good amount of purified recombinant protein with typical yields of 10 – 20 mg per liter culture.
We then characterized our recombinant CaBD by performing similar experiments using fluorescence spectroscopy except that an excitation wavelength at 295 nm was used to selectively excite Trp residues. Ca2+ binding to CaBD enhanced the fluorescence ~40 %, as determined by the integration of the spectra (all subsequent cited enhancements were from integration) and the maximum peak was shifted from 340 nm to 344 nm (Fig. 1A). The calcium-induced fluorescence change indicates that our protein was functionally active, but the enhancement was somewhat lower than the previous report [10]. The reason for this discrepancy is not clear, but it may be due to the difference in the protein constructs such as the different additional N-terminal residues and a longer polypeptide we used (residues 1-184), excitation wavelengths, and/or purification procedures. We also detected significant fluorescence change upon Mg2+ binding though the enhancement was lower (~ 10 %). The signal leveled off after approximately 600 μM of Mg2+, but it is not clear whether all EF-hands were completely occupied at this concentration, mainly due to lack of information regarding the relationship between structural and spectroscopic changes. Therefore, 2 mM Mg2+ was used in our all subsequent experiments except chemical unfolding experiments to ensure saturation. The EF-hands of CaBD appeared to bind specifically to Ca2+ because the addition of 40 μM Ca2+ to Mg2+-bound CaBD quickly converted it to its Ca2+-bound form judged by the different fluorescence profiles of the metal-free and metal-bound forms. This finding is consistent with the previous result showing that Mg2+ does not affect Ca2+ binding to CaBD [10].
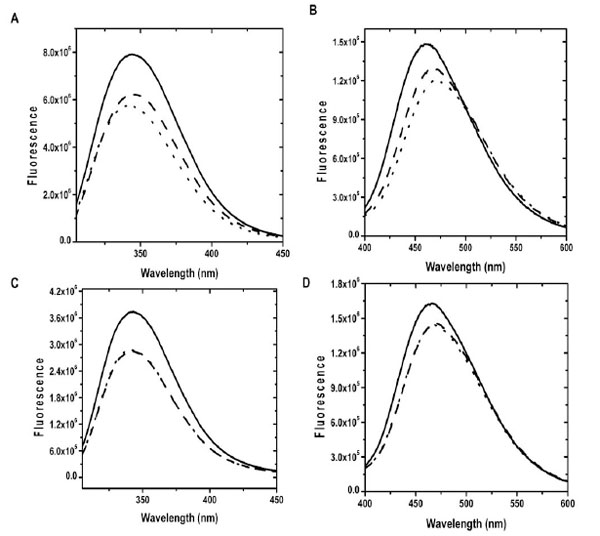
The emission spectra of proteins were recorded at 20 °C in the presence of 500 µM EDTA (dotted line), 2 mM Mg2+ and 1 mM EGTA (dash line) and 100 µM Ca2+ (solid line). (A) and (B) show Trp and AEDANS fluorescence of CaBD, where (C) and (D) show Trp and AEDANS fluorescence of E99Q/E143Q.
Since the intrinsic fluorescence measurements only reflect the changes near the vicinities of the three Trp residues, it is difficult to differentiate the fluorescence contribution from each individual residue. In order to obtain more specific structural information, we labeled the Cys107 residue with IAEDANS to generate AEDANS-CaBD. The Cys107 residue is located between the 3rd EF-loop (residues 88-99) and 4th EF-loop (residues 132-143) in the C-terminal half, and the labeling on this site should serve as a particular probe for the behavior of the C-terminal lobe. Ca2+ and Mg2+ binding to AEDANS-CaBD enhanced the fluorescence intensity by 18 % and 10 %, respectively, and their maximum emission wavelengths were shifted from 470 nm to 466 and 460 nm for Mg2+ and Ca2+ binding, respectively, which suggest the local environment of AEDANS is more hydrophobic in the Ca2+-bound form than in the Mg2+-bound form (Fig. 1B). In our case, fluorescence spectra of Mg2+-CaBD were identical in the presence and absence of EGTA, a specific Ca2+ chelating agent, indicating such signal enhancement upon Mg2+ addition was not due to calcium contamination. Despite the fact that such a change can be monitored by fluorescence spectroscopy, the lack of a direct stoichiometric relationship for the amount of Ca2+ or Mg2+ binding that truly affects Trp and AEDANS fluorescence prevents us determining Ca2+/Mg2+ binding affinity. The increases in Trp and AEDNAS fluorescence caused by Mg2+ binding were smaller than those caused by Ca2+ binding, which indicates the local environments of Trp and Cys107 residues were different among the apo, Ca2+- and Mg2+-bound forms.
ANS Binding and Stern-Volmer Quenching
The dye ANS was used to investigate the solvent-accessible hydrophobic surface in the apo-, Ca2+- and Mg2+-bound states (Fig. 2). Similar to the previous result using TNS [10], Ca2+ binding to CaBD caused ~ 4 fold increase in ANS fluorescence, but Mg2+ binding to CaBD slightly increased ANS fluorescence. These data indicate that Mg2+-CaBD has a structure similar to the apo form. To further test this hypothesis, we determined the accessibility of the Trp residues and the AEDANS motif by fluorescence quenching. Fig. (3) shows the Stern-Volmer plots for the acrylamide quenching. Intrinsic fluorescence quenching revealed an upward quenching curve, which was more obvious with high concentration of quencher (data not shown). The upward curves are consistent with ground state heterogeneity and occurrence of dynamic and static quenching, but it is difficult to analyze those effects due to lack of structural information. In order to easily compare Trp accessibility, we only determined the quenching in the low concentrations of acrylamide and fitted the data with a linear quenching equation to obtain the Stern-Volmer quench constant (Ksv). The structures of the individual forms can be differentiated by fluorescence quenching: Trp residue(s) were more accessible for quenching in the calcium-bound form with the Ksv value of 9.49 M-1, than the Mg2+-bound form with the Ksv value of 5.04 M-1 and the apo form with the Ksv value of 5.28 M-1 (Table 1). AEDANS fluorescence quenching shows a similar trend, but the Mg2+-bound form appeared to be more sensitive than Trp quenching. This suggests that Mg2+ binding causes structural changes in the C-terminal half, but the change does not induce exposure of hydrophobic patches for ANS binding or enhance Trp fluorescence quenching. Both intrinsic and extrinsic fluorescence quenching revealed that there is an increase in the exposure of chromophores upon Ca2+ binding, whereas Mg2+ only affects the C-terminal half. It is not clear whether the N-terminal half binds Mg2+ because there is no suitable fluorophore for detection of Mg2+ binding at that half domain.
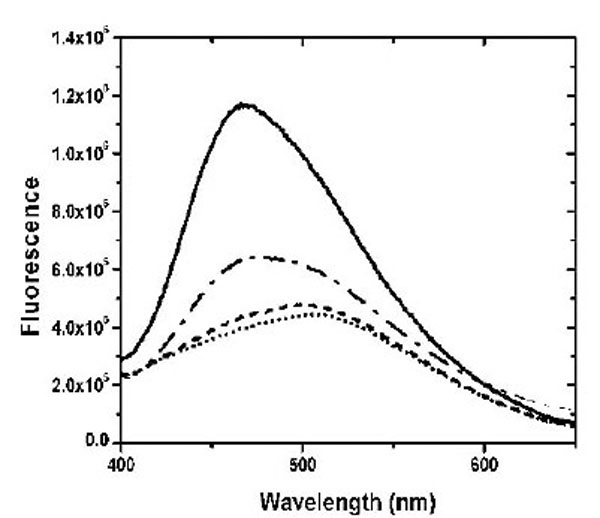
ANS binding to CaBD was monitored in the presence of 500 µM EDTA (dotted line), 2 mM Mg2+ plus 1 mM EGTA (dash line), and 100 µM Ca2+ (solid line). ANS binding to E99Q/E143Q in the presence of 100 µM Ca2+ is presented as a dash-dotted line.
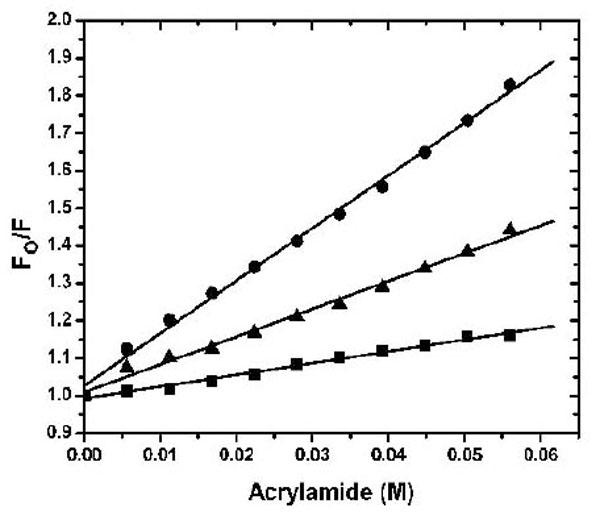
2 µM AEDANS-CaBD in the presence of 100 µM EDTA (square), 2 mM Mg2+ plus 1 mM EGTA (triangle), or 100 µM Ca2+ (circle) was titrated with increasing amounts of acrylamide. The standard Stern-Volmer equation was used to fit the data as described in the experimental section and their Ksv values are shown in Table 1.
Stern-Volmer Quenching Constants Obtained from Acrylamide Titration
a The data were obtained by fitting with a linear Stern-Volmer equation.
b The labeled proteins were excited at 340 nm and their intensities at the maximum emission wavelength were recorded.
Secondary Structures Determined by Circular Dichroism
The studies using fluorescence spectroscopy suggest significant conformational changes in CaBD upon Ca2+ binding. To further determine such change, we used CD spectroscopy to monitor the secondary structure content. In the absence of Ca2+, the CD spectrum shows two negative elliptical peaks near 208 and 222 nm and one large positive band at 190 nm (Fig. 4), which are characteristic peaks of proteins containing a dominate α-helical secondary structure. However, neither the addition of Ca2+ nor Mg2+ significantly changed the CD signal. These data are similar to the studies showing that the secondary structures of the CaM superfamily are nearly unaltered by Ca2+ or Mg2+ binding, but that the angle and distances between structural motifs change [17].
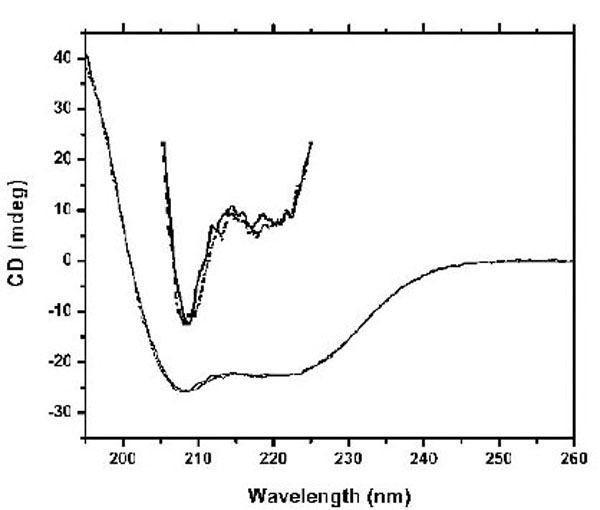
Spectra were recorded at 20 °C in 10 mM Tris, pH 7.5 in the presence of 500 µM EDTA (dotted line) and 100 µM Ca2+ (solid line).
Kinetics of Calcium and Magnesium Association and Dissociation
We used fluorescence spectroscopy coupled with a stopped-flow device to measure the kinetics of the calcium and magnesium association and dissociation of CaBD. To measure calcium association, a solution containing CaBD in the presence of 10 μM EDTA was rapid-mixed with an equal volume of a buffer containing Ca2+ ranging from 50 to 200 μM. The calcium binding was then monitored by fluorescence. In all cases, the calcium binding was too fast to be detected in our stopped-flow spectrometry (data not shown). This fast binding was confirmed by an experiment including ANS in the reactant syringes in the same condition, in which ANS fluorescence increased and reached the maximum at 75 ms (data not shown), thus indicating a slow ANS binding after the fast calcium binding. To trigger the calcium dissociation from CaBD, a large excess amount of EDTA was mixed with Ca2+-CaBD. The kinetic traces obtained fit well to a single-exponential equation (Fig. 5B) and the dissociation rate constant (koff) was determined to be 4.5 ± 0.3 s-1 (mean ± SD, n = 5). The measured kinetic trace was identical to the experiment using AEDANS-CaBD performed in the same condition, indicating that dye-modification did not impair calcium binding, or at least calcium dissociation. However, the amplitude of the fluorescence decay was approximately 55 - 65% of total Trp or AEDANS intensity changes (i.e. difference between the Ca2+- and apo-CaBD), in which the initial intensity for the slow decay was estimated by extrapolating the fitted curve to the time before mixing (i.e. -20 ms), thus implying that there was a non-detectable fast decay. Since AEDANS was labeled specifically in the C-terminal half, it is reasonable to conclude that the slow phase represents the calcium dissociation from the C-terminal half, whereas the fast phase is associated with calcium dissociation from the N-terminal half, in which the cys107-labeled AEDANS is able to sense such a change. To test this hypothesis, we generated a double mutant, E99Q/E143Q, in which the calcium binding sites in the 3rd and 4th EF-hand motifs were removed. We changed the most frequently occurring negative residue, glutamate, at position 12(-Z) to glutamine in its canonical EF-hand motifs because several calcium binding proteins demonstrate that such mutation impairs the calcium binding. Such mutational changes were expected to have minimal overall alternation on protein structure [18]. The mutant’s partial inability to bind Ca2+ was proved by fluorescence quenching in which its Ksv values were almost identical in all states (Table 1), and by the chemical stability discussed below (Fig. 6). Using the unlabeled and labeled E99Q/E143Q, the slow fluorescence decay previously observed in the wild type CaBD disappeared, displaying only a non-detectable fast fluorescence decay (Fig. 5A). Using the dissociation constant for the C-terminal half, Kd,C = 4 μM and the equation Kd,C = koff,C/kon,C, the association rate constant (kon,C) is calculated to be 1.1 x 106 M-1 s-1. If we assume that the fast decay is within the dead time (i.e. 20 ms), the dissociation rate constant for the N-terminal half, koff,N, is estimated to be > 300 s-1, therefore giving kon,N > 1.5 x 107 M-1 s-1 using Kd,N = 20 μM. The lower Kd value for Ca2+ binding to the N-terminal half is mainly from its fast calcium dissociation. The data obtained from the previous study examining binding affinity [10] and our kinetics on CaBD is very similar to CaM, in which its C-terminal half was shown to have a tighter binding affinity and a slower calcium dissociation [19].
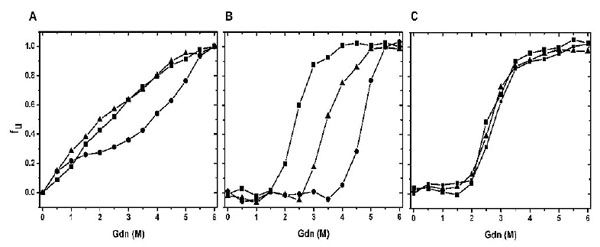
Chemical Stabilities of CaBD and E99Q/E143Q. Proteins were incubated at different concentrations of Gdn, and their maximum wavelengths were recorded. The percentage of unfolding was calculated as described in the experimental section. (A) shows the chemical unfolding of CaBD in the presence of 500 µM EDTA (square), 10 mM Mg2+ (triangle), and 1 mM Ca2+ (circle). (B) and (C) show the chemical unfolding of AEDANS-CaBD and AEDANS-E99Q/E143Q in the same condition. The curves are the average of three independent experiments each.
While the decay of Trp fluorescence can be easily explained due to the relative changes of the two half domains, the decrease in AEDANS fluorescence, implied in the first calcium dissociation (~ 40 % of amplitude), does suggest a significant interaction between the dye and the N-terminal half, possibly through spatial contact and/or the interdomain link. This argument was supported by the fact that the fluorescence spectrum of E99Q/E143Q showed that this mutant still responded to Ca2+ binding with 28 and 9 % enhancements (40 and 18 % for the wild type) for Trp and AEDANS fluorescence, respectively (Fig. 1C & D). The intrinsic fluorescence change for this mutant might be due to the relative change of two rigid halves, i.e. Ca2+-N-CaND and apo-C-CaBD, by which the interface formed through such interaction maintains a weaker ANS binding (Fig. 2). Such an explanation could not be simply applied to AEDANS-E99Q/E143Q. It has been seen that similar mutations on the 1st or 2nd EF-hands in the N-half domain of CaM alter structure, and thus fluorescence, resulting in Ca2+-binding to the C-terminal half domain of CaM, but not Ca2+ binding itself [20]. Furthermore, the possibility of the C-terminal half of this mutant still retaining calcium binding capacity in our experimental condition was eliminated because same mutation on C-CaBD (residues 79-184) resulted in the losing of AEDANS fluorescence response upon Ca2+ binding (unpublished results). Therefore, the calcium binding strengths of the N-terminal half and the C-terminal half are independent as reported previously [10], but their spectroscopic behaviors and thus their structures, appear not to be completely independent. Due to cross communication between the two half domains, our results also raise the question of whether calcium binding within and between the half domains is non-cooperative as previously reported [10].
The kinetics of Mg2+ association was very similar to that of Ca2+. The Mg2+ association was too fast to be detected. The dissociation of Mg2+ triggered by EDTA addition was only observed using AEDANS-CaBD (Fig. 5C). The dissociation rate constant was determined to be 6.65 ± 1.02 s-1. The amplitude of such decay was identical to the intensity change determined from the steady states, which suggests that this Mg2+ dissociation is from the C-terminal lobe. This conclusion is supported by the fact that AEDANS-E99Q/E143Q did not respond to Mg2+ even at very high concentration of Mg2+. Interestingly, the Mg2+ dissociation from CaBD was too fast to be detected by Trp fluorescence.
Chemical Stabilities of CaBD and Mutant
Since the data from the previous result indicates structural independence of the N- and C-terminal halves, the stability of the individual domains may be different in relation to their thermodynamics and kinetics. We therefore performed chemical unfolding induced by guanidine hydrochloride (Gdn). Fig. (6A) shows a plot of the fractions of unfolded protein as a function of Gdn concentrations. The unfolding of CaBD monitored by intrinsic fluorescence was not a two-state unfolding model. This is consistent with the fact that the three Trp residues are near the peptide ends where structures tend not to be compact. In the presence of Ca2+, the overall stability was enhanced. There were two obvious transitions at ~ 1 and 4.5 M Gdn with an intermediate at 2-3 M Gdn. The calcium-bound CaBD is substantially more stable than the apo form, and the result agrees with the prediction from the theory of ligand binding [21]. Unfortunately, there is no constructive information that can be gathered from intrinsic fluorescence. To acquire specific structural stability, the same experiment using the cys107-labeled CaBD was conducted. Increasing Gdn concentration resulted in red shift in wavelengths from 470 to 496 nm in the absence of Ca2+ (data not shown); the unfolding curve, corrected for denaturant effects, appeared to be a two-state model. Fig. (6B and 6C) show a plot of the fraction of unfolded protein as a function of denaturant, and the parameter Cm was determined at the concentration of Gdn where 50% of the protein was induced unfolded. The unfolding monitored by the extrinsic fluorescence gave the Cm values approximately 2.5 M and 4.7 M in the absence and presence of Ca2+, respectively; the latter being consistent with the second transition obtained from intrinsic fluorescence. The data also indicated that the Cm values of the apo forms of N-terminal and C-terminal halves were near 2.5 M. Ca2+ binding most likely increases the stability of the C-terminal half, but not the N-terminal half. This conclusion was supported by using AEDANS-E99Q/E143Q. The unfolding curves were identical in the presence of EDTA, Mg2+, and Ca2, consistent with our previous fluorescence quenching result showing inability of calcium binding in the C-terminal half. The unfolding study of Ca2+-bound AEDANS-E99Q/E143Q also suggests that the Cm value for Ca2+-bound N-terminal half is ~2.5 M. Interestingly, Mg2+ appeared to stabilize CaBD, more specifically the C-terminal half. This unfolding is concentration-dependent (data not shown) because lowering Ca2+ or Mg2+ concentration destabilizes CaBD as seen in CaM [21]. It appears that the domain that has a higher calcium binding affinity has a higher degree of stability. This is seen for the C-terminal half, in which its higher chemical stability is due to a higher unfolding free energy difference (ΔGNU) between the apo and calcium-bound states. In contrast, the N-terminal half domain has free energies which are comparable in apo and calcium-bound forms, thus lowering ΔGNU. More thorough unfolding studies with the individual halves using appropriate labeling or other techniques will further determine their free energy difference and cooperative unfolding transition for each domain. Our result indicates that the correlation between binding affinity and domain stability for CaBD agrees with other calcium binding proteins [20, 22].
CONCLUSION
Ca2+-induced electron transfer controlled by the CaBD-flavoprotein interaction in NOX5 is very important in the regulation of its superoxide production. In fact, we found that the heme reduction (Fe3+ to Fe2+) of NOX5 was only observed in the presence of Ca2+ in an anaerobic condition (Wei and Stuehr, unpublished result), suggesting that this domain-domain interaction mediates electron transfer from NADPH to FAD and/or FAD to heme. Previously Bánfi et al. had generated GST-fused CaBD and demonstrated its conformational change upon Ca2+ binding mainly by Trp and TNS fluorescence [10]. We extended such studies by characterizing CaBD and the E99Q/E143Q mutant. Our finding is similar to what they reported on Trp fluorescence enhancement and hydrophobic patch formation upon Ca2+-binding. However, we investigated more thoroughly on such conformational change by including acrylamide quenching, chemical stability, and secondary structure change by CD spectroscopy. The kinetics of Ca2+ association and dissociation was studied by stopped-flow spectrometry. Our data indicated that the C-terminal half of CaBD has a higher Ca2+ binding affinity, a higher chemical stability, and a slow Ca2+ dissociation. By using site-specific AEDANS labeled proteins, our data also suggest the N- and C-terminal halves of CaBD are structurally dependent.
Furthermore, Mg2+ binding to CaBD was observed in the intrinsic and extrinsic fluorescence. Whether Mg2+-CaBD is of any biological importance is not known. But like other EF-hands containing proteins, Mg2+ may play a role in a cellular condition, where its concentration is 0.5-2 mM and Mg2+ may either occupy two or all four calcium binding sites. The exchange of Ca2+ for Mg2+ is likely to limit the association and dissociation of the calcium ions [23]. Indeed, the rate of Mg2+ dissociation for CaBD was comparable to calcium dissociation. In resting cells, NOX5 appears to bind Mg2+ and its structure mostly resembles the apo form. As calcium concentration elevates upon cell stimulation, the rate of Ca2+ association to CaBD is compromised by a slow dissociation of the bound Mg2+ ions. The chemical unfolding study suggested that such Mg2+ binding enhances the stability of CaBD in resting cells. Mg2+ binding only alters the local conformational changes, particularly near the C-terminal end, while Ca2+ binding leads to global conformational changes of the two half domains.
Recent studies indicate that NOX5’s Ca2+ sensitivity is enhanced when its flavoprotein domain interacts with CaM [13] or is phosphorylated at its Thr494 and Ser498 residues [14]. Since the activation still relies on Ca2+ binding, exactly how the structural change in the C-terminal flavoprotein end induced by the above mechanisms propagates to CaBD remains to be studied further. Therefore, structural information about CaBD gathered from spectroscopy is important for investigation of such structural communication.
ABBREVIATIONS
ACKNOWLEDGEMENTS
We thank Dr. Carl Frieden at Washington University at St. Louis for allowing us to use his CD spectrometer. We thank the financial support from Cottrell College Science Awards (CCSA) of Research Corporation and Summer Research Fellowships (SRF) from Southern Illinois University Edwardsville (SIU Edwardsville) to CCW; Research Grants for Graduate Students (RGGS) from SIU Edwardsville to LQC and TJ; and Undergraduate Research Academy (URA) from SIU Edwardsville to NM, LH, and KL. We also thank Kristen Wetherell, Nicole Reynolds, and Tiffany Williams (SIU Edwardsville) and Anjali Shekar (O’Fallon High School, supported by 2009 Summer Students and Teachers As Research Scientists (STARS)) for helping with protein expression and purification.
REFERENCES
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]

