All published articles of this journal are available on ScienceDirect.

Analysis of Endothelial Protein C Receptor Functionality on Living Cells’
Abstract
Activated protein C (APC) is a major control system of blood coagulation. APC prevents coagulation pathway by degrading Va and VIIIa plasma’s coagulation factors. Protein C activation requires its binding to specific endothelial cell receptor (EPCR). APC binding to EPCR also activates a wide range of defense mechanisms (anti-inflammatory, antiapoptosis…). EPCR expression by cells can be detected by various methods, including immunoanalysis and molecular biology. However, no assays evaluate its functionality. A method, inspired of a standard fibrinoformation time assay, was developed to estimate EPCR ability to bind APC on living cell surface in vitro. Endothelial cells were incubated with APC and fibrinoformation on cells was followed by spectrophotometry (plasma absorbance increases with fibrin polymerization). Membrane-bound EPCR retain APC, thus prolonging fibrinoformation time in a dose-dependent manner. Control was realized with EPCR-negative cells. This new method can be used on any cell type to study the expression of other coagulation receptors.
INTRODUCTION
Bleeding time is widely used as a screening test for primary hemostasis or platelet-vessel interaction and, as such, is a measurement of intrinsic function. Hemophilia diagnosis was originally based on the presence of a prolonged whole blood clotting time.
The transformation of soluble fibrinogen into insoluble fibrin is a crucial step of thrombus generation and requires the presence of phospholipids (as a component of platelet) and calcium. The partial thromboplastin time (PTT) is the fibrinoformation time of plasma mixed with exogenous phospholipids (cephalin) and calcium chloride [1]. The test is termed "partial" due to the absence in the reaction mixture of tissue factor, the major cellular initiator of blood coagulation. The addition of kaolin, a non-specific surface activator, shortens the coagulation time and makes it much more reproducible [2]. Compared to PTT, this method is named activated PTT (aPTT). Since its introduction, aPTT spread rapidly throughout the clotting community. PTT/aPTT are nowadays standard screening tests employed in clinical laboratories for the diagnosis of coagulation deficiencies.
This test actually remarkably optimizes research on the anticoagulant pathway and elucidation of thrombophilia pathogenic mechanisms [3]. Increasing numbers of aPTT test systems are nowadays commercially available and they differ in chemical composition of the reagent, activator and the method of endpoint detection [4,5]. In these systemes, the coagulation process is mainly followed by spectrophotometry since plasma absorbance increases with fibrinoformation.
Besides aPTT important conceptual role in our understanding of hemostasis in vitro, this method had major impact on elucidation of activated protein C (APC) resistance underlying molecular mechanisms [6-8]. APC acts as a major control system for thrombosis through negative feedback of fibrin generation. Synthesized by the liver as a zymogen, protein C (PC) binds its receptor on endothelial cell surface (endothelial protein C receptor or EPCR) in order to be activated. Activated protein C together with its cofactor prevents coagulation pathway by degrading two factors essential for clotting to occur (FVIIIa and FVa) [9-13]. Moreover, the binding of APC to its receptor EPCR activates a wide range of defense mechanisms in the host such coagulopathic and anti-inflammatory reactions [14], calcium flux [15], metalloproteinase production [16], and anti-apoptosis [17,18] to name just a few.
EPCR expression by cells can be detected by various assays, including immunocytochemistry, molecular biology or soluble EPCR quantitative immunodetection. However, to our knowledge, no assays evaluate EPCR functionality in situ. A method, inspired of aPTT, was developed to estimate EPCR ability to bind APC on cell surface [19]. Cells were incubated with APC and fibrinoformation of a plasma added onto cells was evaluated by spectrophometry analysis. APC trapped on endothelial membrane, correlated to the amount of EPCR expressed by those cells, would increase plasma-clotting time.
MATERIALS AND METHODS
Reagents
Plasma
Blood samples were provided from the haematology department, at the Hotel Dieu hospital (Paris, France). Blood was anticoagulated in buffered sodium–citrate and plasma were obtained after 30min centrifugation (2000g). All samples were frozen at -80°C until they were analysed. To avoid any bias due to haemostasis factors abnormalities, a minimum of 5 different plasmas were pooled and homogenised for each experiment.
Cells
Human endothelial bone marrow (HBMEC) cell line, provided from Dr Pienta, [20] was cultured in 75-cm culture dishes containing Endothelial Basal Medium (EBM) supplemented with 10% fetal bovine serum (FBS), 5 mM L-glutamine, 200 IU/ml penicillin and 10 µg/ml streptomycin (PAA Laboratories Inc, Etobicoke, ON, USA) according to the recommendations of the supplier. The human breast adenocarcinoma cell line MCF7 was grown in RPMI 1640, as described above.
Various reagents were obtained from different sources as follows: human recombinant APC (Lilly, Suresnes, France), PTT-LA reagent which contains a phospholipid (cephalin) and a particulate activator (micronized silica) in a buffered medium, and CaCl2 (generously provided by Diagnostica Stago).
RT-PCR Analysis
Human endothelial cells RNA extracts were prepared using the Nucleospin RNA-II kit (Macherey-Nagel EURL, Hoerdt, France). Briefly, five million bone marrow endothelial cells were washed with ice-cold PBS and lysed with a specific buffer that prevents RNA degradation and enhances RNA adsorption to the silica membrane of the filtration column. DNAse I was then directly applied to the membrane to digest contaminating DNA. After several washes, pure total RNA was finally eluted with RNase-free water.
One microgram of total RNA was used for reverse transcription into the complementary DNA (cDNA). RNA was denatured for 5 min at 65°C and incubated 30 min at 42°C in a final volume of 20 µL in an appropriate buffer (Tris HCl pH 8.8 5 M, (NH4)2SO4 2 M, MgCl2 1 M) with 1.25 mM dNTP (Sigma), 10 pmol oligo(dT) (Promega) and 200 units of M-MLV reverse transcriptase (Gibco, Paisley, UK). A control was performed without reverse transcriptase. The mixture was heated to 72°C for 3 min at the end of the reaction.
The polymerase chain reaction (PCR) was performed on transcribed cDNA, using specific primers of EPCR genes (sense: 5'-CAACTTCAGGATGTTGACAA-3'; antisense: 5'- CTACAGCCACACCAGCAAT-3'). These synthetic primers were designed with Primer3 program, BLAST verified and synthesized by Eurobio (Les Ulis, France). The reaction mixture consisted of 15 ng cDNA, 2.5 units Taq DNA polymerase (Gibco), 200 mM dNTP, 0.2 mM of the respective oligonucleotide primers, 10 mM Tris-HCl, pH 8.3, 50 mM KCl and 1.5 mM MgCl2 in a final volume of 50 µl. The mixture was amplified in a thermal cycler (GeneAmp 9600, Perkin Elmer, Wellesley, USA). Denaturation was carried out at 94°C for 1 min, followed by annealing step at 55°C for 1 min and an extension step at 72°C for 1 min to a total of 35 cycles. Controls without reverse transcriptase or cDNA or using beta2-microglobulin primers were similarly amplified in parallel. The PCR products (15 µL) were electrophoresed on 1.2% agarose gels (Gibco) in 0.45 mM Tris-borate containing 0.2% ethidium bromide and 1 mM EDTA. The gels were photographed along with a 100 bp DNA ladder, which had been run concurrently as a size marker.
Immunocytochemistry
Five thousand HBMEC were grown in glass bottom chamber slides (Lab Tek, Nunc, Naperville, IL, USA) and rapidly fixed and permeabilized with ice-cold methanol. After three washes with PBS, the non-specific binding was saturated by exposure to 10% bovine serum albumin (BSA) in PBS for 15 min. The cells were thereafter incubated for 1 h at room temperature with 20 µg/ml of EPCR specific antibodies (AF2245, R&D Systems, Minneapolis, MN, USA), and then washed with PBS. The slides were then incubated successively with appropriate biotinylated secondary antibodies and streptavidin fluorescein (Amersham, Buckinghamshire, UK). Isotypic control was performed and cells nuclei were DAPI-labelled. Bound fluorescein was directly observed on a Nikon fluorescence microscope. For FACS analysis, endothelial cells were detached using accutase (PAA) and immunolabelled, without preliminary fixation, as previously described. A negative control was performed using goat IgG1 isotype control (Southern Biotech, Birmingham, AL, USA). EPCR expression was analysed with FACS Calibur.
Functionality Assay
Activated partial thromboplastin time (aPTT) is a test measuring clotting time in plasma, leading to evaluate intrinsic clotting pathway efficiency. In order to activate this pathway, phospholipid and a particulate activator (PTT-LA) and calcium are mixed with a plasma sample. The time is measured until thrombus formation. To confirm APC efficiency to increase aPTT of normal plasma, 20 µL APC at different concentrations (dilution in serum-free medium) were added to mixed plasma and PTT-LA. Clot formation kinetics was then compared between plasma samples with and without APC.
A method, inspired of the aPTT, has been developed to estimate the expression by any type of cells of EPCR, the receptor of the anti-thrombotic molecule activated protein C. Cells were incubated with APC and clotting time of plasma added onto cells was measured. Membrane-bound EPCR would retain APC which degrade coagulation factors Va and VIIIa, thus increasing plasma clotting time.
Thirty thousand HBMEC were seeded on a 96-well plate (tissue-culture test plate, TPP, Trasadingen, Switzerland) and incubated overnight with appropriate culture medium. The day after, cells were washed and incubated with APC (1 to 50 µg/mL, 15min at 4°C). Controls were performed with serum-free medium. Cells were then washed repeatedly with the culture medium devoid of serum, and further incubated with normal plasma and PTT-LA (100 µL each, 3 min at 37°C). PTT-LA reagent contains a phospholipid (cephalin) and a particulate activator (micronized silica) in a buffered medium. CaCl2 (25 mM) was finally added to the mix to trigger coagulation. The optical density (OD) of each well was determined by a spectrophotometer at 37°C (Wallac Multilabel Counter 1420, PerkinElmer, Waltham, MS, USA). APC incubations were realized at 4°C to avoid any putative receptor internalization whereas clotting assays were performed at 37°C, temperature suitable for haemostasis enzymes activity. Several wavelengths were tested (from 355 to 595nm). A measure was achieved every minute and plasma aPTT in presence of cells incubated with APC was compared to aPTT realised with untreated cells. Results are expressed as clots formation kinetics.
RESULTS AND DISCUSSION
To certify the endothelial phenotype of the HBMEC cell lines used as a model, expression of some typical markers (vWF, E selectin, VEGF R-2 and EPCR) were assessed by gene array, PCR and immunocytochemistry (results not shown). To ensure the synthesis of endothelial protein C receptor (EPCR) mRNA by the endothelial cell lines, a RT-PCR using specific primers of EPCR was performed on HBMEC. The corresponding electrophoresis gel presented an amplification band at 692 bp, consistent with the size predicted, whereas water control was negative (Fig. 1, panel A). Expression of EPCR was confirmed by immunocytochemistry, using specific antibodies (Fig. 1, panel B).
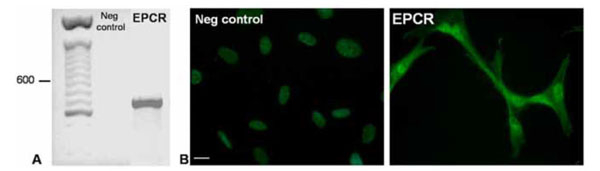
(A) EPCR amplification was performed on HBMEC cDNA, using specific primers. An expected band at 692bp was observed. A negative control, realized without cDNA, was run concurrently (panel A, second lane). DNA ladder is displayed in the first lane.
(B) EPCR proteins were detected on endothelial cells by immunocytochemistry. Cells were grown in glass bottom chamber slides, rapidly fixed and successively incubated with EPCR specific antibodies (20 µg/ml) and appropriate FITC-coupled secondary antibodies. Isotypic control was performed and nuclei were DAPI-labelled (first picture of panel B). Bound FITC was directly observed on a fluorescence microscope (initially x60, scale bars represent 10 µm). Big white arrows show nuclei, little arrows show cell extension.
Increasing number of automated coagulation analysers, using turbidimetric and absorbance technologies, are commercially available. However standard 405 nm light filter permits various coagulation and fibrinolysis tests, various wavelengths have already been tested to assess clot formation (350nm [21]; 570nm [22]).
Optimal wavelength to study fibrinoformation was thus determined as follow. Plasma, PTT-LA and calcium were mixed in a 96-well plate and clot formation kinetics were analysed at different wavelengths (Table 1). Optical densities vary a lot from plasma to another, it seemed thus important to select a wavelength that confers to plasma a lowest absorbance to reduce these discrepancies. The choice of 595 nm was finally made even though an upper absorbance range could have discriminated more efficiently unclotted and clotted state.
Optical Density of aPTT mix, Before and After Coagulation, Obtained at Different Wavelengths
| Wavelenght (nm) | O.D. Unclotted | O.D. Clotted | Ratio |
|---|---|---|---|
| 355 | 0,446 | 1,385 | 3,11 |
| 405 | 0,493 | 1,365 | 2,77 |
| 490 | 0,267 | 0,989 | 3,70 |
| 540 | 0,115 | 0,767 | 6,67 |
| 595 | 0,076 | 0,642 | 8,45 |
Plasma and PTT-LA (100 µL each) were incubated 3min at 37°C in a 96-well plate. CaCl2 (100µL) was added to the mix to trigger coagulation. Clot formation was followed by spectrophotometer at various wavelengths. A measure was realized every minute. This table represents absorbance of the mix before and after fibrinoformation. The ratios (O.D. clotted/O.D. unclotted) of these absorbancies are displayed in the last column.
APC efficiency to increase aPTT of normal plasma was then confirmed. APC induces a dose-dependent aPTT prolongation, as shown on Fig. (2). Kinetic obtained with 0.1µg/mL APC was similar to the control (results not shown), which ensures the reproducibility of the method. Literature recommends an APC concentration range of 1-100 µg/mL to detect its anticoagulant effects or intracellular properties [23,24]. Our findings are in agreement with these previous works.
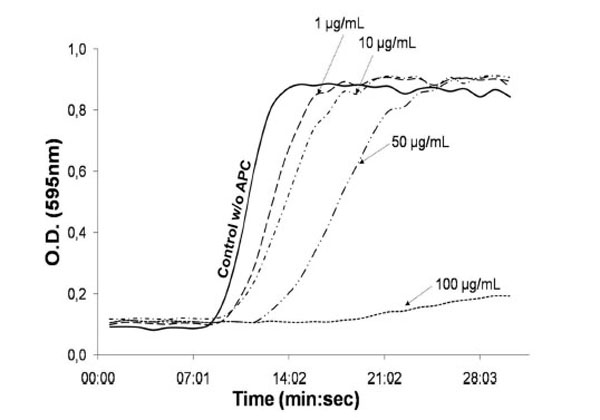
Plasma, PTT-LA (100 µL each) and 20 µL APC (from 0.1 to 100 µg/mL) or serum-free medium (control w/o APC) were incubated 3min at 37°C. CaCl2 (100 µL) was added to trigger the coagulation. Fibrinoformation was followed by spectrophotometer. Absorbance was measured every minute.
Endothelial cells were then seeded on a culture plate and exposed to APC for 15 min. The incubation was performed at 4°C to avoid EPCR internalization. APTT was then performed onto cells carefully washed after exposure to different levels of APC. The coagulation time shift induced by APC is highly appreciable and exhibits a dose-dependent response (Fig. 3A). Data in Fig. (3B) also indicate that 5 min incubation with 10 µg/mL APC is sufficient to increase aPTT. Nevertheless, a longer incubation was chosen to optimize the experiments.
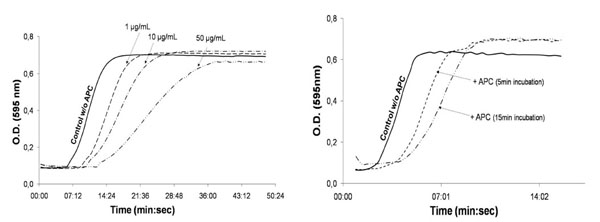
30,000 endothelial cells were seeded per well in a 96-well plate. When confluency reached 80%, cells were incubated at 4°C either 15 min with increasing dose of APC (A) or with 10 µg/mL APC or serum-free medium (control w/o APC) for 5 or 15 min (B). Cells were thereafter carefully washed and incubated (3min, 37°C) with plasma and PTT-LA (100 µL each). CaCl2 (100 µL) was finally added to trigger the coagulation. Fibrinoformation was followed by spectrophotometer. Absorbance was measured every minute.
Finally, a dose-dependent aPTT was achieved with epithelial cells that do not express EPCR as demonstrated by FACS analysis (Fig. 4A). The aPTT was realised with the procedure previously described. As expected, no shift in the coagulation time was observed after APC incubation (Fig. 4B). This result ensures that the cells were carefully washed after APC exposure and that the fibrinoformation delay was indeed due to EPCR ability to retain APC on the cell membrane.
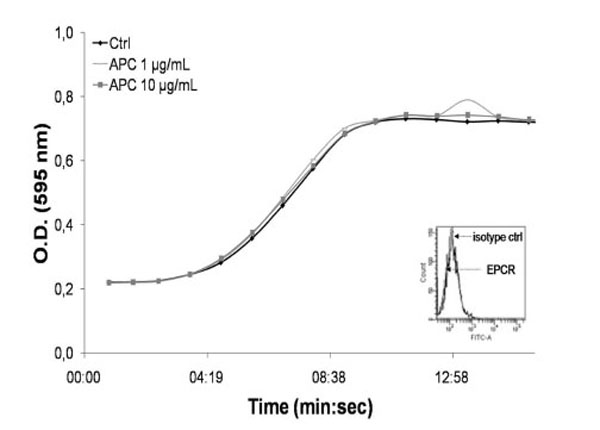
Epithelial cells were detached using Accutase and immunolabelled as previously described with EPCR antibodies or isotypic control. EPCR expression was analysed with FACS Calibur. The FACS cytogram is displayed in the bottom right of the panel.
30 000 epithelial cells were seeded per well in a 96-well plate. When confluency reached 80%, cells were incubated with APC (1 or 10 µg/mL) or serum-free medium (control w/o APC) for 15 min, at 4°C. Cells were thereafter carefully washed and incubated (3min, 37°C) with plasma and PTT-LA (100 µL each). CaCl2 (100 µL) was finally added to trigger the coagulation. Fibrinoformation was followed by spectrophotometer. Absorbance was measured every minute.
APTT remains a standard hemostatic screening test daily used throughout the world in clinical and research laboratories. This easy and reproducible assay can furthermore evolve with study needs. APTT provides indeed an interesting tool for long-term anticoagulant therapy control [25], for detection of fetal lung maturity with the use of amniotic fluid [26] or even for gene transfer monitoring in hemostasis disorders [27].
While number of soluble EPCR quantitative determination immunoassays already exist [28, 29], this aPTT-based method was designed to assess EPCR presence and functionality on cellular membrane. Contrary to classical aPTT, this new assay can take in account whole cells.
The present assay is suitable to estimate cellular EPCR expression and functionality since a significant and dose-dependent fibrinoformation time shift is observed after APC incubation of endothelial cells. No delay is observed with EPCR-negative cells, which ensures that fibrinoformation time extension is imputed to APC attached to the cell membrane via EPCR. This method was initially developed to assess EPCR expression and functionality but could be easily extended to study the expression by any cell type (underneath or included into a clot) of any coagulation factors’ receptors. This aPTT-based assay could also assess global coagulant activity of cells. This could be efficient to study thromboembolic disorders as hypercoagulability that is frequently associated with cancer.
ABBREVIATIONS
REFERENCES
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]

