All published articles of this journal are available on ScienceDirect.

Differential Activity of Caspase-3 Regulates Susceptibility of Lung and Breast Tumor Cell Lines to Paclitaxel
Abstract
Recent development of tumor resistance to paclitaxel presents a major problem to cancer treatment. An unsettled controversy in the cancer chemotherapy field, however, is whether caspases play a prominent role in paclitaxel-induced death in tumors. Previous studies suggest that cleavage of caspase-3 is not instrumental for the execution of death in tumors treated with paclitaxel, while other reports indicate that caspase-dependent pathways may be critical for paclitaxel cytotoxicity. In this study, we investigated the role of caspase-3 in breast and lung tumor cell line sensitivity to paclitaxel. Clonogenic survival and live/dead viability-assays, together with enzymatic activity and cell proliferation assays, reveal that the levels of paclitaxel-induced caspase-3 enzymatic activity in tumor cells correlate directly with tumor sensitivity to the drug.We observed a 2-fold increase in caspase-3 activity in 4T1-Luc breast tumor cells, but a 3-fold and 4-fold decrease in A549 and A427 lung tumor cell lines, respectively. Together, our results suggest that caspase-activation and activity levels are not only key determinants of paclitaxel-induced death in tumors but also serve as good indicators for tumor susceptibility to paclitaxel therapy. Our studies also indicate that within clinically relevant doses of paclitaxel, the ability to rid tumor populations of dormant tumor cells controls the rate of tumor recurrence.
INTRODUCTION
More than thirty-eight years after its isolation from the bark of the Pacific Yew tree, paclitaxel has become the first clinically relevant representative of a novel class of anticancer drugs used to treat a wide variety of patients suffering from breast, lung, head, neck, and ovarian cancer [1-10].However, recent development of paclitaxel resistance by tumor cells is worrisome and unsettling for clinicians since this affects efficacy of therapy and presents a major obstacle for survival of cancer patients. Drug resistance by tumors also complicates the development of potent cancer treatments. Although current knowledge of the factors contributing to drug resistance phenotype has increased significantly [11,12], the roles of downstream targets of cell death, such as caspase 3 & 7, in tumor sensitivity to paclitaxel therapy have not been fully explored. Given their requisite role in apoptosis, caspases are potentially attractive targets for cancer treatment. Elucidating the factors regulating caspase activation is therefore crucial to understanding how tumors manipulate these pivotal death effectors and to identify novel molecular targets and strategies for improving current therapy.
In this study, we report a framework in which differential activity of caspase-3 in lung versus breast tumor cell lines controls tumor sensitivity and resistance to paclitaxel. We found that lung tumor cells, with repressed caspase-3 activity, were more resistant to paclitaxel than breast tumor cells. Thus, our findings point to an underappreciated role of caspases in tumor resistance to drug and support other studies suggesting that targeting caspase activity is crucial for improving the potency of chemotherapeutic agents.
MATERIALS AND METHODS
Cell Culture
The human non-small cell lung carcinoma lines, A549 & A427 were obtained from American Type Culture Collection (Manassas, VA, USA) and maintained in basic medium as described [13]. In addition, mammary tumor cell line, 4T1-Luc, was obtained from American Type Culture Collection (Manassas, VA) and maintained as described [14].After growing to 100% confluence, cells were trypsinized (0.5 mL trypsin in 5 mL PBS) and incubated for 5 -10 min. The cells were then harvested by low speed centrifugation, washed in 1 mL media and seeded at 3 X 104cells per 200 µL of medium per well in a 96-well plastic plate (Nunc Inc. Rochester, NY). A portion of cells (1 X 105 cells/well) was also grown overnight on Lab Tek 8-chamber slides (Nunc Inc. Rochester, NY) for caspase-activity and live/dead assays.
Cell Proliferation Assay
The effect of paclitaxel on tumor cell growth was evaluated using Cell Titer 96®Aqueous One Solution Cell Proliferation Assay (Promega). This assay permits the detection of metabolic activity of proliferating cells without the required solubilization steps in the MTT assay. Briefly, 24 h drug-treated cells were cultured in a 37˚C humidified incubator for 2 h with 20 µL of CellTiter96®Aqueous One Solution Reagent containing tetrazolium compound [3-(4, 5-dimethyl-thiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium salt; MTS] and an electron coupling reagent, phenazine ethosulfate (PES). The absorbance per well was measured at 490 nm using a micro-plate reader (Perkin Elmer Wallac Victor™ 1420 Multilabel Counter).
Live-Dead Viability and Clonogenic Survival Assays
Cell viability and integrity after drug treatment was assessed by two assays. With the live-dead viability kit (Invitrogen), drug-treated cells were incubated with ethidium homodimer (EthD-1) for the last 45 min of the drug exposure period. After this treatment, the media was removed and slides were prepared for microscopy by mounting with prolong gold antifade® reagent (Invitrogen, Molecular Probes). For clonogenic survival assay, drug-treated cells were cultured in a 37˚C humidified incubator for 10 days to allow colony formation. Drug exposure times varied from 2 to 14 days for time course study. Cells were then fixed with methanol, stained with solution containing 20% methanol and 0.5% crystal violet, and clusters made up of 100-cells were counted as surviving colonies. The percentage of surviving colonies relative to untreated cells was determined by the proportion of colonies produced from cells after drug treatment. Plating efficiency was 80% ± 10%.
Caspase-3 Activity Assay
To quantify the levels of basal and induced caspase-3 protein and corresponding activity in different tumor cell lines, we used a fluorometric enzymatic assay, EnzChek® Caspase-3 (Molecular Probes, Eugene, OR). Cell extracts from paclitaxel-treated (25 µM) and untreated A549, A427 and 4T1-Luc were treated with final substrate concentration of 200 μM and incubated at room temperature for 1 h. Fluorescence was measured with a micro plate reader using excitation at 360± 40nm, and emission at 460 ± 40 nm. Appropriate controls were performed to determine background fluorescence of the substrate. A standard AMC-substrate curve was used to quantify the levels of enzyme-cleaved substrate from the measured fluorescence values.
Statistical Analysis
Statistical analysis was performed using the Student’s t-testand nonlinear regression analysis in GraphPad Prism Software® (version 4.0 for windows, San Diego, CA).
RESULTS
High Concentrations of Paclitaxel Delineate the Differential Susceptibility of Non-Small Cell Lung Carcinoma and Metastatic Breast Cancer Cells to Chemotherapy
Studies of the human non-small cell lung carcinoma cell lines, A549-T12 and A549-T24, show that while these paclitaxel resistant cells are cultured and maintained in nanomolar levels of drug, the parental A549-cell line from which they are derived is, in contrast, sensitive to nanomolar drug doses [15-20]. To test the ability of nanomolar- concentrations of drug to inhibit cell proliferation, we investigated the viability of the A549 and A427cell lines after exposure to 1- 1000 nM of paclitaxel using a modified MTT assay [19,21].We observed that 24 h treatment of A549 and A427-cells with concentrations ≤ 200 nM did not impede cell viability. Cells showed a minor response at concentrations ≥ 500 nM, with A427 being slightly more susceptible than A549-cells in the 500 t0 1000 nM range (Fig. 1A). Overall, proliferation of either cell line was not affected by nanomolar concentrations of paclitaxel. However, increasing paclitaxel doses to the micromolar scale remarkably decreased proliferation of A549 and A427 cells. The half maximal effective concentration (EC-50) for these cells was determined to be 22.5 μM and 12.5 μM, respectively (Fig. 1B and C). In addition, we measured the response of the 4T1-luc cell-line to equivalent micromolar drug concentrations. In contrast to A549 and A427-cells, 4T1-luc cells showed a strong dose response with EC-50 of 8.5 μM (Fig. 1D). At concentrations >40 μM, all three cell lines succumbed to death without distinct behavioral differences. However, disparate susceptibility of the three cell lines was pronounced at a dose range of 10 – 25 μM. While the anti-proliferative effect of paclitaxel was enhanced in 4T1-Luc cells, A427 and A549 cells were relatively less affected by the drug (Fig. 1E).
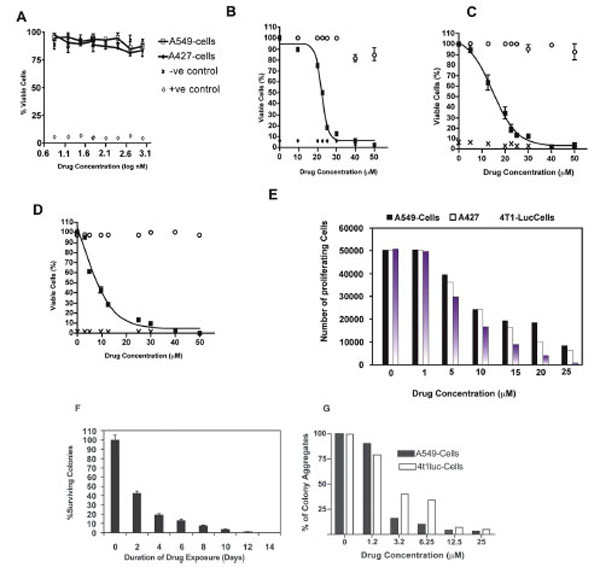
Cell viability was determined by a modified MTT assay. After 24-h drug exposure with indicated concentrations, cells were incubated with 20 µl of MTS reagent for 2-h. Absorbance values were measured at 490 nm. Experiments were done in triplicate for each cell type, with appropriate controls. The half maximal effective concentration (EC50) of paclitaxel for each cell type was determined by nonlinear regression analysis using GraphPad Prism Software® (version 4.0 for windows, San Diego, CA). Data represent normalized mean values. (A) Nanomolar concentrations of paclitaxel have little effect on tumor cell viability especially at lower concentrations. However a dose response is observed in both tumor cell types, with A427 cells showing slightly increased sensitivity than A549-cells at higher nanomolar drug concentrations (500 nm and 1000 nm).Open circles represent negative control, crosses and diamonds represent positive control, black rectangles represent samples. (B) EC50 for A549 cells was determined to be 22.5 µM. (C) EC50 for A427 was determined to be 12.5 µM and (D) EC50 for 4T1-Luc was determined to be 8.5 µM. At high drug doses > 40 µM, no significant difference in tumor cell line response to paclitaxel is observed.However, clear differences in cell type sensitivity to drug dosage is observed at concentrations < 20 µM. Of the 3 tumor cell lines,A549 and A427 show low sensitivity to the drug at low concentrations, with 4T1-Luc cells appearing to be the most sensitive in this dose range. (E) Anti-proliferative effect of paclitaxel is enhanced in 4T1-Luc cells, but not in A427 and A549 cells at high drug doses. Proliferation of tumor cells after 24-hour drug treatment was analyzed by MTS assay (Molecular Probes), as described in methods section. Experiments were done twice in triplicates. N=6. At concentrations ≥ 10 µM, a drastic reduction in the number of proliferating cells was observed in 4T1-luc, in comparison with A549 and A427 cells which showed significant cell proliferation. (F) 4T1-luc tumor cells show increased aggregated and clustered colony formation. Clonogenic survival assay was performed to evaluate the behavioral response of tumor cells to an extended duration of drug exposure. A time course study was performed to determine colony formation at a fixed drug concentration (12.5 μM) over varying exposure times (2- 14 days). Each data point was derived from the results of at least three independent experiments. N= 9. Data show mean ± SEM of three experiments, each in triplicate. Relative to untreated controls, a time dependent-decline in the number of colonies formed was observed in 4T1-luc cells, but not in A549 or A427 cells (not shown). In these latter cell lines, cells were more dispersed and very few aggregated colonies (cell clusters consisting of more than 100-cells) were observed. (G) 4T1-luc cells form colony aggregates in a dosedependent fashion, relative to A549-cells which show reduced cell colonies at higher drug concentrations. Overall, however, more proliferating A549 cells than proliferating 4T1-luc cells were present at higher concentrations.
The relatively higher number of persistent proliferative cells in A549 and A427-cells suggests that both tumor cell lines potentially have an intrinsic resistance to paclitaxel. To further assess the behavioral response differences between the lung and breast tumor cell lines, we performed clonogenic survival assays, where tumor cells were treated with varying concentrations of the drug for an extended period (2-14 days). A time dependent-decline in the number of 4T1-luc cells, relative to the untreated controls, was observed (Fig. 1F). Although all three cell lines showed survival dose response, 4T1-luc tumor cells formed more aggregated and clustered colonies. In contrast, A549 and A427 cells appeared more dispersed, with less aggregated colonies (i.e. one or two clusters made up of more than 100-cells) (Fig. 1G).This suggests that 4T1-luc cells perhaps respond more to prolonged drug therapy by forming aggregates while A549 cells appear to resist paclitaxel treatment by spreading over the plate surface.The differences in chemotherapeutic response among the 3 cell lines clearly point to the underlying differences in their resistance mechanisms to paclitaxel [22-24].
Paclitaxel Induces Distinct Death Phenotypes in Breast and Lung Carcinoma Cell Lines
Although it is fairly well known that most tumor cells undergo paclitaxel-induced apoptotic death, it is equally well established that paclitaxel induces other non-apoptotic programmed cell death mechanisms in different tumor cell types [20,21,25]. To ascertain and distinguish between apoptotic and non-apoptotic death, we used a Live/Dead® Viability assay to evaluate the quintessential cellular features of death such as plasma membrane blebbing, DNA fragmentation, and cell shrinking. We particularly examined drug-treated cells for the presence of fragmented nuclei and compromised plasma membranes as indicators of death. We observed a dose-dependent fragmentation of nuclear DNA as well as damage to the cell membrane. This phenotype was highly pronounced in 4T1-luc tumor cells with extensive fragmentation at 50 μM and 25 μM of drug, visualized as red punctate dots in dead cells stained by EthD-1 (Fig. 2, A-F). Very few live cells were observed at these concentrations and cells had a mix of necrotic and apoptotic characteristics. Nonetheless, the extent of membrane disintegration and nuclear damage in 4T1-luc cells was more moderate at 12.5 μM and 6.25 μM of paclitaxel (Fig. C & D). Like 4T1-luc cells, A549 cells showed dose-dependent DNA fragmentation, but with less damaged cell membranes (Fig. 3, A-F). Moreover, dead cells had fewer and less dispersed punctate nuclear fragments (Fig. 3A, insert). Interestingly, we observed a few mitotic cells among the apoptotic A549 cell population, which were immobilized at the mitotic phase in the presence of paclitaxel. (Fig. 3A, D & F). The absence of this distinct population of cells in 4T1-luc cells clearly highlights different death response mechanisms in the two tumor cell lines. Previous studies reported that paclitaxel suppressed microtubule dynamics and disrupted the functioning of the mitotic spindle [26-29], suggesting a slow cell cycle progression at the metaphase/anaphase checkpoint that eventually induces cell death. The features in A549-cells fit perfectly with this pattern, albeit, we also observed a mix of apoptotic morphologies in this cell line. Our results demonstrate that 4T1-luc breast tumor cells respond to paclitaxel by initiating extensive nuclear fragmentation and inducing both necrotic and apoptotic forms of death. On the other hand, paclitaxel-induced death in A549 cells occurs partly through interference with cell cycle progression, thus, pointing to different phases of drug-induced apoptosis in the two tumor cell lines.
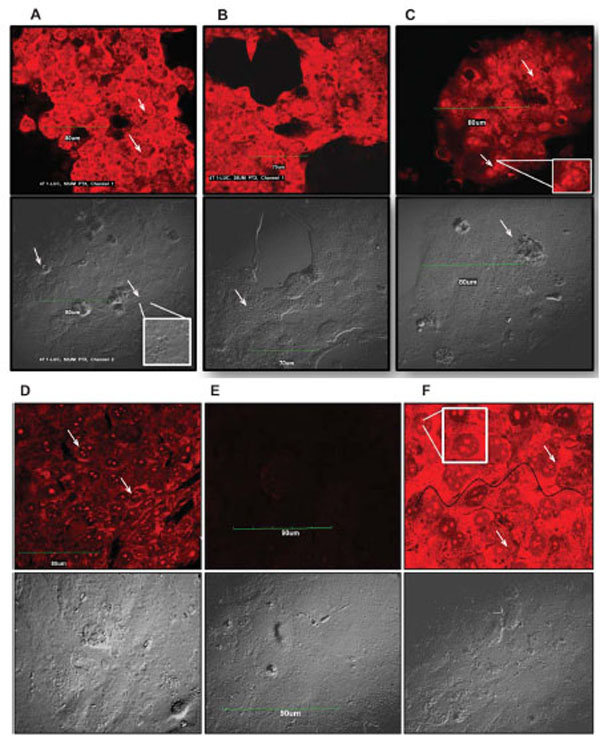
The extent of DNA damage observed in tumor cells was dose dependent. Arrows point to fragmented nuclei. Inserts highlight nuclear fragments and damaged DNA within cells. Scale bar indicates 90 µm. Cell morbidity was determined by Live-Dead Viability Assay. Drug treated cells were incubated with ethidium homodimer (EthD-1), which stains fragmented nuclei of dead cells. EthD-1 dye can only penetrate cells with compromised membranes and emits red fluorescence (ex/em - 495 nm/ 635 nm) on binding to nucleic acids. Exclusion of the dye by intact membrane of live cells distinguishes dead cells from live ones. (A – F) - Apoptosis in 4T1-Luc cells. Cells treated with high paclitaxel doses, 50 µM (A) and 25 µM (B), show extensive nuclear fragmentation and damaged membranes. (C) At 12.5 µM and (D) 6.25 µM, moderate nuclear fragmentation is observed. (E) Reflects background fluorescence from negative control cells. (F) Apoptotic induction in positive control (ethanol-treated) cells. Wavy black line traces boundary of damaged cell membrane.
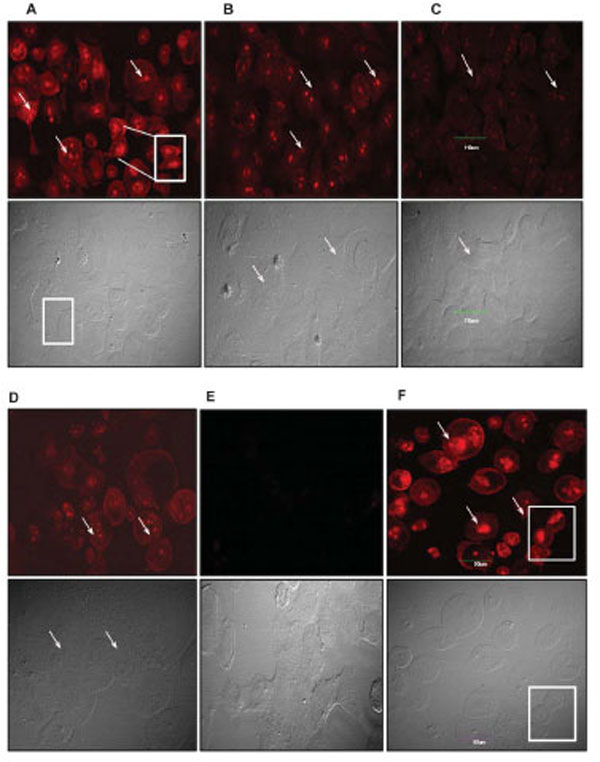
Arrows point to fragmented nuclei. Inserts and boxed regions highlight fragmented nuclear DNA and mitotic cells. These distinct populations of mitotic cells were not observed in 4T1-luc tumors. Scale bar indicates 90 µm. Cell death was determined by Live-Dead Viability Assay as described. Exclusion of EthD-1 dye by intact membrane of live cells distinguishes dead cells from live ones. Images are shown in red and grey channels. (A – F) -Cell death in A549 cells. Fragmented nuclei are visible in cells treated with high paclitaxel doses, 50 µM (A) and 25 µM (B), as well as in cells treated with moderate doses, 12.5 µM (C) and 6.25 µM (D). (E) No dead populations are observed in untreated cells; mitotic cells are visible in the grey channel. In contrast to the morphology of 4t1-luc tumor cells, mitotic proliferating A549 cells may contribute to A549 tumor resistance at low doses of paclitaxel. (F) Apoptotic induction in positive control (ethanol-treated) cells.
Paclitaxel Induces a 2-Fold Increase in Caspase-3 Activity in 4T1-Luc Cells but a 3-Fold and 4-Fold Decrease in A549 and A427 Tumor Cell Lines, Respectively
Based on reports that somatic mutations of caspase-3 gene in human cancers cause selective inactivation of cysteinyl proteinases to compromise apoptotic death [28-31], we hypothesized that differences in the intrinsic and drug-induced activity of effector caspases contribute to the acute paclitaxel-induced death response in 4T1-luc versus the benign response in A549 and A427 tumors. We predicted that 4T1-luc cells would have relatively higher basal and drug-induced caspase-3 activity than A549 or A427 tumor cells. We found that while all 3-lines showed some levels of basal enzymatic activity, A427-cells exhibited the least activity with 40 µM of cleaved substrate, followed by A549-cells with 54 µM of cleaved substrate, and then 4 T1-Luc cells, with 68 µM of substrate (Fig. 4A). A decrease in activity in the presence of a caspase-3 inhibitor corroborated the specificity of the reaction. On measuring caspase-3 activity in extracts from drug-treated cells, we found increased activity in 4T1-luc cells but a decrease in A549 and A427-cells (Fig. 4B). This suggests that while 4T1-luc cells activate caspase-3 on exposure to drug, A549 and A427 cells somehow repress caspase-3 activation. In contrast to the drug-treated 4T1-luc cells that showed a two-fold increase in caspase-3 activity in comparison to basal activity levels, A549 and A427 showed approximately a 3-fold and 4-fold decrease in enzymatic activity, respectively, relative to basal levels (Fig. 4C). This result provides evidence that increased activation of caspase-3 confers sensitivity of 4T1-1uc cells to the drug, whereas repressed caspase-3 activation potentially accounts for the resistance of A549 and A427 cells to paclitaxel.

The intrinsic and drug-induced caspase-3 activity of 3-cell lines were determined by a fluorimetric enzymatic assay that measures cleavage of a caspase-3 specific substrate, Z-DEVDAMC,where Z represents a benzyloxycarbonyl group, and AMC, 7-amino-4-methylcoumarin. The substrate fluoresces weakly in the UV range (Ex/Em~330/390 nm) but yields strong blue-fluorescent product (Ex/Em ~ 342/441 nm) upon proteolytic cleavage and allows continuous monitoring of the activity of caspase-3 and its related proteases in cell extracts. A standard AMC-substrate curve was used to quantify the levels of enzyme-cleaved substrate from the measured fluorescence values. To confirm that the observed fluorescence signal was due to the activity of caspase-3-like proteases, control treatments were incubated with 10 M of a reversible caspase-3 inhibitor, Ac-DEVD, prior to treating cells with the substrate. Experiments were performed twice in triplicate. N= 6. Data show mean ± SEM of 2 experiments done in triplicate.
(A) While all 3-lines showed some level of basal enzymatic activity, A427-cells showed the least activity with 40 µM of cleaved substrate, followed by A549-cells, with 54 µM of cleaved substrate. 4 T1-Luc cells showed the highest levels of activity with 68 µM of cleaved sub-strate. A decrease in activity in the presence of a caspase-3 inhibitor confirms the specificity of the reaction. The underlying difference in caspase-3 activity may account for the observed resistance patterns after drug exposure. (B) Paclitaxel induces differential caspase-3 activity in tumor cell lines. Whereas 4T1-Luc cells showed an increase in levels of cleaved product (120 µM, 60% of added substrate) after exposure to drug, A549 and A427-cells showed a decrease in the levels of cleaved product, with 20 µM and 11 µM, respectively, of cleaved substrate. This suggests that while 4T1-luc cells activate caspase-3 on exposure to drug, A549 and A427 cells somehow repress caspase-3 activation. (C) In comparison to basal enzymatic activity levels, paclitaxel induces a two-fold increase in caspase-3 activity in 4T1-luc cells as measured by increased levels of cleaved substrate. In contrast, A549 and A427 show approximately a 3-fold (2.7) and 4-fold (3.64) decrease in enzy-matic activity, respectively. This increased activation of caspase-3 underlies the sensitivity of 4T-1uc cells to drug, whereas, repressed cas-pase-3 activation potentially accounts for the resistance of A549 and A427 cells to paclitaxel
DISCUSSION
Research on paclitaxel-resistance in A549 tumor cell lines reveals that the P-glycoprotein efflux pump, the known mediator of multi-drug resistance phenotype, plays no major role in resistance in this cell line [30,31]. In addition, although paclitaxel-induced tubulin gene mutations and alterations in total tubulin content have been reported to limit tumor cell cycle progression, these observations give a partial account of how tumors overcome drug cytotoxicity [32,33]. Moreover, an unsettled controversy in the cancer chemotherapy field is whether caspases feature prominently in paclitaxel-induced non-apoptotic and apoptotic forms of death and what the implications are for tumor cell resistance to paclitaxel [2,23,25,34-37]. We explored this issue further by determining whether the basal and drug-induced activity levels of caspase-3 underlies the differences in susceptibility of breast and lung tumor cell lines to paclitaxel.
Our study reveals that, relative to the lung tumor cell lines examined, the breast tumor cells are less resistant to drug, and require about 2.5 fold less drug to reach the half-maximal effective concentration for killing cells. In contrast to studies emphasizing the disruptive effect of nanomolar concentrations of paclitaxel on microtubule dynamics in tumor cells [15,16], we observed no significant changes in either breast or lung tumor growth at nanomolar drug concentrations. However, at micromolar levels of paclitaxel treatment, clear differences in breast and lung tumor sensitivity nanomolar scale were evident. While nanomolar concentrations of drug may impact tubule dynamics and mitosis, the differences in our findings suggest that the global effects on tumor survival at such low drug concentrations are less threatening to the tumor cells. In preclinical trials, detectable plasma concentrations of paclitaxel is dependent on the initial dose, mode of delivery and pharmacokinetics, and have varied from 0.05 nM to 0.1 μM, after IP paclitaxel microspheres treatment [38].
Although it has been argued that over time, cells treated with nanomolar concentrations of paclitaxel accumulate intracellular drug levels to the micromolar scale [16], our results highlight crucial differences in the temporal and behavioral response of tumors bombarded immediately with micromolar levels versus tumors experiencing a gradual increase in drug levels. In the former scenario, the marked sensitivity of tumors to drug suggests that a sudden influx of drug at high concentrations affords tumors little time to initiate a counteractive response. However, in the latter scenario, the absence of a pronounced effect on tumor growth suggests that within the time window where intracellular concentrations build up to micromolar levels, tumor cells are able to recoup many drug resistant processes to combat the toxic effects of paclitaxel. Thus, breast and lung tumor cells evade death at low paclitaxel concentrations.
The apparent difference in the phenotypic outcomes of low versus high drug concentrations has significant implications for cancer therapy. For one, it delineates the importance of using micromolar drug concentrations within the clinically achievable plasma concentrations (<10μM) for therapy [2,38-41]. These high concentrations probably limit the time window in which tumor tumors are able to regroup and resist drug action. High drug concentrations also afford distinctions in tumor susceptibility to drugs and enhance the potential to kill dormant cancer cells that are hibernating in cell cycle arrest within tumor populations. A note of caution here, however, is that it is of clinical importance to study whether the use of higher drug concentrations aggravates sides effects such as inflammation and neurotoxicity.
The variation in chemotherapeutic response of breast and lung carcinoma cell lines clearly point to underlying differences in their resistance mechanisms to paclitaxel. While previous studies using colon, B-lymphoma and lung cancer cells have argued against the key role of effector caspases in paclitaxel-induced cytotoxicity [17,25], our findings evoke a direct correlation between caspase-3 activation and tumor susceptibility to paclitaxel. The observation that lung tumor cells which are more resistant to paclitaxel exhibit as much as a four-fold decrease in caspase-3 activity points to an underappreciated role of caspases in tumor resistance to drug. In contrast to other reports in support of caspase-independent response to paclitaxel at nanomolar levels and different tumor cell lines [25,42], the positive response found in this study could be attributed to the use of micromolar levels of drug and breast/lung tumor cells. Although nanomolar levels of paclitaxel did not allow good delineation of tumor susceptibility, they induced different tumor behavioral responses in cells than do high drug concentrations. Our results also support the concept of tumor cell line specificity of caspase-regulated resistance to drug.
An interesting challenge arising from this study relates to deciphering factors that account for the differential activity of caspases in different tumor lines. The differences in cleavage mechanisms of caspases in different tumors or the presence of endogenous inhibitors preventing caspase-3 activity in resistant tumor cell lines are probably involved. Some studies have shown that constitutive levels of known endogenous inhibitors of caspases, such as XIAPs, do not correlate with paclitaxel resistance patterns in tumors [35,43]. Combined with our study, it is obvious that other mechanisms or yet unidentified inhibitors of caspase-3 play a key role in preventing enzyme activation in some tumor cells.
CONCLUSION
In summary, the functional status of caspases plays a key role in the response of tumors to drugs. Specifically, nanomolar concentrations of paclitaxel may not be optimal for killing tumors but provide invaluable information about caspase-mediated response of different tumors to the drug. Even in tumor cell lines that subsequently develop resistance to treatment, the activity levels of caspases determines susceptibility of the tumors to paclitaxel. Considering that all the tumor cell lines examined responded positively at micromolar concentration, a delicate balance between side effects and benefits of the drug at high concentrations is needed to optimize cancer cell death. The potential to use tumor-specific drug carriers to increase the local concentration of drugs in tumors relative to healthy tissues would improve treatment outcome.
ACKNOWLEDGEMENTS
We thank Bao Xu, Dr. Duanwen Shen, and Dr. Sharon Bloch for valuable suggestions and discussions. This study was supported in part by NIH grants ((R01 CA109754, R01 EB1430, R33 CA100972, and R21 CA123537)
REFERENCES
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]

