All published articles of this journal are available on ScienceDirect.

The Optimization of Soluble PTEN Expression in Escherichia coli
Authors Info & Affiliations
Abstract
As a vital tumor suppressor, PTEN (Phosphatase and tension homolog deleted on chromosome 10) is involved in inherited syndromes, and is among the most frequently inactivated tumor suppressor gene in sporadic cancers. PTEN loss-of-function widely occurs in human cancers via a variety of mechanisms, including genetic alterations and posttranslational modification. These suggest PTEN has a role of functional importance in a variety of cancers. In the present study, we constructed a prokaryotic expression vector that efficiently expresses GST-PTEN (the target protein in which PTEN is fused with glutathione S-transferase tag) in E. coli. We found that the target protein was partially soluble although major portions of the protein remained in the inclusion bodies. Furthermore, we explored the optimal induction temperature, isopropyl β-D-1-thiogalactopyranoside (IPTG) concentration and induction time in a series of experiments. Expression level analysis indicated that PTEN reached its peak level at 36○C for 8 h with 1.5625mM IPTG, while solubility analysis revealed the optimal induction temperature was at 20○C, the optimal IPTG concentration was 0.1µM and the optimal induction time was up to 8 h. Taken together, we provide an optimal induction condition for expressing soluble fusion protein of PTEN in E. coli, facilitating further analysis of PTEN’s biological function in vitro.
Introduction
PTEN was originally identified as tumor suppressor frequently lost or mutated in various human malignances [1, 2]. PTEN loss-of-function widely occurs in human cancers via a variety of mechanisms, including genetic alterations and dysfunction [3].
PTEN is a 403 amino acid and dual lipid/protein phosphatase which can convert second messenger PIP3 (phosphatidylinositol 3,4,5-triphosphate) to PIP2 (phosphatidylinositol 4,5-bisphosphate), thus repressing the activity of PI3K/AKT signaling associated with cell proliferation, apoptosis and survival [4]. PTEN has also been reported to exhibit protein phosphatase activity: in vitro studies have shown that it can catalyze the dephosphorylation of protein substrates at Ser/Thr and Tyr residues [5]. Genetic alterations or dysfunction of PTEN causes neurological defects and various human cancers [6].
Structurally, PTEN is composed of phosphatase domain, C2 domain and C-terminal tail. The phosphatase and C2 domains make up the catalytic unit which converts PIP3 to PIP2, contributing to the tumor suppression. The C-terminal tail plays an important role in regulation of PTEN phosphatase activity [7], thus mutations in this region leads to tumorigenesis [8].
PTEN, once considered a strictly cytoplasm-located protein, is now known to be present in the nucleus [9]. The mono-ubiquitination of PTEN leads to its nuclear importing and stability, modulating genomic stability, growth suppression, oxidative damage and tumorigenesis [10]. Also, SUMOylation of PTEN regulates its nuclear importing [11]. Previous study had demonstrated the fact that the nuclear PTEN can command the chromosome stability through two different mechanisms: (i) PTEN maintains centromere stability through direct interaction with centromeres; (ii) PTEN modulates DNA repair through transcriptional regulation of Rad51, a key protein involved in double-strand break repair [12, 13].
Recent studies have identified a secreted PTEN protein referred to as PTEN-long. It was reported to have an alternative start code site as translation of PTEN-long. Compared to classical PTEN, the alternative PTEN protein has additional 173 amino acids containing secretion signal peptide added at its N-terminal, which allows the protein to be secreted into the extracellular environment and to enter in recipient cells, thereby inhibiting the PI3K-Akt pathway in a paracrine fashion [14-16].
To investigate PTEN’s biological function in vitro, a soluble PTEN protein or fusion protein with a tag is required. Glutathione S-transferase (GST) is originally found in eukaryotic, and this unit is usually fused with target protein and used in eukaryotic express systems. The application of GST tag is essential because it is soluble and facilitates solubility of the fusion protein. Moreover, the target protein can be purified by binding to the immobilized glutathione (as substrate of GST) beads [17, 18]. In the present study, PTEN with GST tag was cloned in pGEX-6p-1 vector to explore the expression conditions to obtain soluble GST-PTEN. We have optimized induction conditions to increase the solubility of GST-PTEN fusion protein, which provides an important approach to analyze interaction between PTEN and novel candidate proteins.
Materials and Methods
Materials
The expression vector pGEX-6p-1 was originally obtained from Amersham (Shanghai China). The pGEM-T-easy vector was purchased from Promega (Shanghai China). The BL21 recipient bacterium strain was purchased from Beijing DingGuo ChangSheng Biotechnology (Peking China). A plasmid mini preparation kit was purchased from Omega Biotek (Guangzhou China). DNA polymerase, Restriction endonucleases, T4 DNA ligase, IPTG, protein molecular weight markers, DNA ladder and DNA Gel Extraction Kit were obtained from TaKaRa (Dalian China). The PCR primers were synthesized by Sangon Biotech (Shanghai China). The ProteinPure GST Resins were purchased from TransGen Biotech (Peking China). The Coomassie Brilliant Blue R250 and BSA were purchased from Sigma (Shanghai China) and the PTEN primary antibody was obtained from Santa Cruz (Shanghai China). The ECL Kit was purchased from Boster (Wuhan, China).
Methods
Construction of GST-PTEN Expression Plasmid
The Human PTEN gene (1212bp, NCBI Reference Sequence NM_000314.4) was amplified by PCR using a plasmid as template which including PTEN coding region conserved in our lab and gene specific upstream primer 5ʹ-GGATCCATGACAGCCATCATCAAAGAG-3ʹ and downstream primer 5ʹ-GAATTCTCAGACTTTTGTAATTTGTGT-3ʹ that included BamHI and EcoRI restriction sites, respectively. The PCR products were subjected to the 1% agarose gel electrophoresis, then purified using an agarose gel extraction kit and cloned into the pGEM-T easy vector. After DNA sequencing, the PTEN coding region was sub-cloned into pGEX-6p-1 GST expression vector and the plasmid construct was confirmed by restriction enzyme digestion. The blank empty plasmid was used as the negative control.
Expression of Recombinant GST-PTEN inE.coli Under Different Inducing Conditions
The successfully constructed plasmid pGEX-6p-1-PTEN was transformed into competent cells BL21 and spread evenly on an LB agar plates (Contents/L: NaCl 10 g, yeast extraction 5 g, tryptone 10 g, agar 15 g) containing 100 μg/ml ampicillin, and then cultured for 14 h at 37○C. Single colonies were selected and incubated into 2 ml of LB medium (Contents/L: NaCl 10 g, tryptone 10 g, yeast extraction 5 g) containing 100 μg/ml ampicillin and cultured overnight at 37○C with shaking at 210 rpm. Subsequently, the overnight culture was diluted into new 15 ml LB medium (at proportion of 1:200) with ampicillin (100 μg/ml) in a 50 ml conical flash and incubated in the 37○C shaker at 210 rpm until it reached an OD600 of 0.6-0.8. Expression of GST-PTEN was induced at different temperatures, IPTG concentration and induction time. To explore the optimum induction temperature for soluble GST-PTEN fusion protein cells were cultured at 20○C, 22○C, 24○C, 26○C, 28○C, 30○C, 32○C, 34○C, 36○C and 38○C induced by 0.5 mM IPTG shaking at 160 rpm for 4 h. To determine the optimal concentration of IPTG for maximal yield of soluble protein, cells were induced with 1x10-4 mM, 5x10-4 mM, 2.5x10-3 mM, 12.5x10-3 mM, 62.5x10-3 mM, 31.25x10-2 mM, and 1.5625 mM IPTG at 20○C while shaking at 160 rpm for 4 h. Lastly, cells were induced with 1x10-4 mM of IPTG for 2 h, 4 h, 6 h, 8 h and 10 h at 20○C.
Cell Lysis and Purification of GST-PTEN Fusion Protein
In the above described cultures, cells from 5 mL culture were harvested by centrifugation at 10,000 g for 1 min and the pellet was resuspended in 400 µl lysis buffer (containing 25 mM Tris, 1 mM EDTA, pH 7.5, 150 mM NaCl, 1 mM PMSF and 0.5% of TritonX-100). The cell suspension was frozen at -80 ○C for 30 minutes and then thawed out on ice. Subsequently, sonication was carried out with 10 % power put out for 30 seconds by 3 times. To separate the soluble protein from the cell debris, the cell lysate was centrifuged at 12,000 g for 10 minutes at 4○C. The supernatant was collected and filtered with 0.22 µm filter before transferring into new tubes. For purification, the 100 µL of ProteinPure GST Resin beads was equilibrated with 1 mL lysis buffer and washed by inverting tube several times and centrifuged at 2500 rpm for 1 minute at 4○C. Combine the supernatant with the equilibrated resin beads and allow to bind at 4○C for 2 h on an orbital shaker. Subsequently, the incubated resin beads were repeatedly washed three times with lysis buffer by centrifugation for 1 minute at 2500 rpm. The sample resin beads were boiled following by SDS-PAGE analysis.
SDS-PAGE and Western-blot Assay
The supernatants and pellets collected from all samples were dissolved in 50 µl PBS to which was added 5×SDS loading buffer. Sample aliquots were boiled, centrifuged and separated on 10% SDS-PAGE gel. Coomassie Brilliant Blue was used to stain the SDS-PAGE gels to determine the solubility profile of the GST-PTEN fusion protein.
To identify GST-PTEN fusion protein, The cells induced with 0.5 mM IPTG and cultured at 26○C for 4 h were subjected to western blot analysis. The PVDF membrane was blocked using 5% BSA in TBST (10 mM Tris, 100 mM NaCl, 0.1% Tween-20, PH 7.6) at room temperature for 1 h, and immuno-blotted with PTEN primary antibody at 1:500 dilution in 4○C overnight. For secondary antibody incubation, the goat anti-mouse IgG was used at 1:10,000 dilution in room temperature for 1 h. Visualization was performed by Enhanced Chemiluminescence using Biospectum Imaging system (Fluorchem E FE0684, Proteinsimple, USA).
Gray Scale Analysis
SDS-PAGE gels were scanned using Epson EXP10000XL. The gray values of target protein bands and total bands at each lane were measured by Adobe Photoshop CC. The graphs were analyzed by GraphPad Prism 5.0 software.
Results
Construction of GST-PTEN Prokaryotic Expression Plasmid
The PTEN coding sequence was first amplified by PCR from a plasmid encoding the full-length PTEN gene (Fig. 1A). The PTEN coding sequence was subsequently inserted into the multiple cloning sites at the BamHI and EcoRI sites of pGEX-6p-1-PTEN expression vector (Fig. 1B). The resulting plasmid pGEX-6p-1-PTEN coded for the recombinant GST-PTEN fusion protein with GST tag at the N-terminal (Fig. 1B). To identify the recombinant plasmid restriction enzymes BamHI and EcoRI were used and a 1212bp and a 4984bp fragment were characteristic and could be separated on a 1 % agarose gel (Fig. 1C). The restriction enzymes digestion assay shows that the pGEX-6p-1-PTEN prokaryotic expression plasmid was constructed successfully. Our next aim was to optimize expression of the protein to yield sufficient amounts of GST-PTEN.
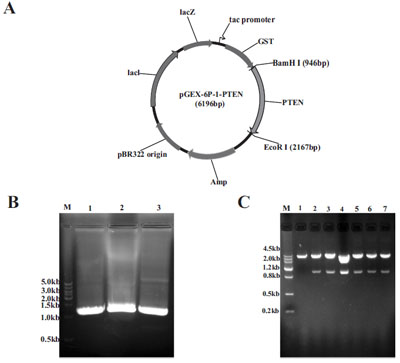
Development of the pGEX-6p-1-PTEN expression vector. (A) PCR products of PTEN encoding region. (B) Map of the pGEX-6p-1- PTEN expression construction and the orientation of the PTEN insertion fragment. (C) Restriction enzyme digestion with BamHI and EcoRI (lane 2-7) of six different clones before transforming into competent BL21 E.coli. The blank empty pGEX-6p-1 vector was used as the negative control (lane 1). DNA Marker (lane M).
The Expression and Solubility Assessment of GST-PTEN Protein Induced at Different Temperatures
Transformed BL21 E. coli induced with 0.5 mM IPTG and cultured at 26○C for 4 h, indicated that target GST-PTEN protein was partially soluble whilst the majority of the protein was insoluble in inclusion bodies (Fig. 2A). The target protein was verified by western blot analysis with anti-PTEN antibody (Fig. 2B). In this study, we used a series of induction temperatures that varied from 20○C to 38○C. The expression level of the target protein (GST-PTEN) did not appear to vary greatly between 20○C to 30○C, increased when induction temperature increases from 32○C to 36○C but then decreases at 38○C (Fig. 3A-D). However, the ratio of soluble to insoluble protein (S % to P %) exhibited a decreasing trend as the induction temperature increased (Fig. 3E). These results suggest that the solubility of GST-PTEN decreases as the induction temperature increased, such that the solubility of GST-PTEN was highest at the low induction temperature of 20○C.
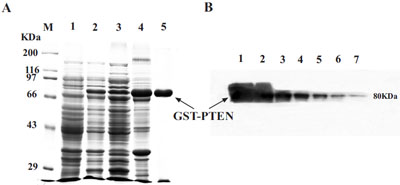
(A) SDS-PAGE assay for the expression and purification of GST-PTEN. Lane M, protein standard markers. Lane 1, pre-induction control; Lane 2, post-induction control; Lane 3, supernatant; Lane 4, pellet; Lane 5, purified GST-PTEN. (B) Verification of GST-PTEN protein by western blot analysis with specific anti-PTEN antibody. Lanes 1-7 are 1:2 serial dilutions of the fusion protein from lane 2 in A. The molecular weight of GST-PTEN is approximately 80 KDa.
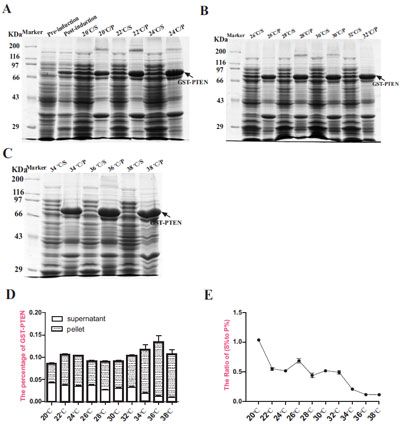
Expression analysis of GST-PTEN induced at different temperatures ranging from 20○C to 38○C. (A, B, C) SDS-PAGE analysis of samples induced with 0.5 mM IPTG for 4 h at different temperatures (S=supernatant; P= pellet). (D) The percentage of target protein (GSTPTEN) to the total protein content. (E) The ratio of (S % to P %) reflected the solubility of GST-PTEN protein (S % = the percentage of soluble GST-PTEN in total protein expressed; P % = the percentage of insoluble GST-PTEN in total protein expressed). Each data point represents the average of three replicate experimental runs.
The Expression and Solubility Assessment of GST-PTEN Induced with Different Concentrations of IPTG
We next sought to determine the optimal IPTG inducing concentration. Proteins from cultures induced with varying concentrations of IPTG were separated by 10% SDS-PAGE and stained by Coomassie Brilliant Blue (Fig. 4A, B). Our results indicate that the expression level of GST-PTEN increased as the IPTG concentration increased and subsequently reached a peak at the maximal 1.5625 mM concentration used (Fig. 4C). However, the ratio of soluble to insoluble protein (S % to P %) decreased with increasing IPTG concentration (Fig. 4D). The data reveal 1x10-4 mM as the optimal IPTG concentration for obtaining maximal solubility of GST-PTEN (Fig. 4D).
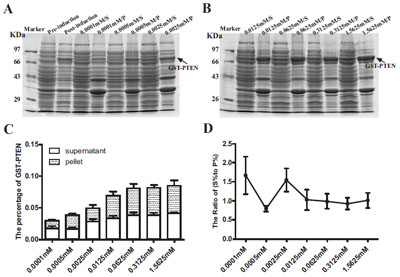
Expression analysis of GST-PTEN induced at different IPTG concentrations ranging from 1x10-4 mM to 1.5625 mM. (A, B) SDSPAGE analysis of samples induced with different IPTG concentrations for 4 h at 20○C (S=supernatant; P= pellet). (C) The percentage of the target protein (GST-PTEN) to the total protein content. (D) The ratio of (S % to P %) reflected the solubility of GST-PTEN protein (S %=the percentage of soluble GST-PTEN in total protein expressed; P %= the percentage of insoluble GST-PTEN in total protein expressed). Each data point represents the average of three replicate experimental runs.
The Expression and Solubility Assessment of GST-PTEN Induced for Different Time Periods
After determining the optimal induction temperature and the optimal IPTG inducing concentration, we next sought to determine the optimal time for protein induction. Induced proteins were separated by 10% SDS-PAGE and stained by Coomassie Brilliant Blue (Fig. 5A, B). The expression of GST-PTEN fused protein increased as the induction time increased and peaked at 8h with a slight decline at 10h (Fig. 5C). This decline in protein production may be due to the increasing burden of bacterial metabolism because of the exotic protein expression. However, the ratio of soluble to insoluble protein (S % to P %) declined as the induction time increased to 6h but subsequently increased at 8h and 10h (Fig. 5D) although this was not significantly different from 4h time period.
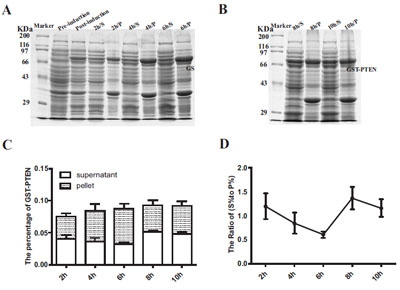
Expression analysis of GST-PTEN induced for different time periods ranging from 2h to 10h. (A, B) SDS-PAGE analysis of culture samples induced with 1x10-4 mM IPTG for different time periods (2h to 10h) at 20○C (S=supernatant; P= pellet). (C) The percentage of the target protein (GST-PTEN) to the total protein content. (D) The ratio of (S % to P %) reflected the solubility of GST-PTEN protein (S %=the percentage of soluble GST-PTEN in total protein expressed; P %= the percentage of insoluble GST-PTEN in total protein expressed). Each data point represents the average of three replicate experimental runs.
Discussion
The pGEX-6p-1-PTEN expression construct was developed by cloning the PTEN gene (coding sequence) into the prokaryotic expression vector pGEX-6p-1 carrying the tac promoter, Lac-operator system and GST sequence, thus we can easily purify PTEN protein by GST tag using GSH resins.
Generally, the temperature, IPTG concentration and induction time have an effect on the intracellular expression of eukaryotic protein in E.coli. Larentis’ research had demonstrated that the pre-induction temperature almost had no influence on the expression of target protein [19]. However, induction temperature has a great impact on protein expression, and the solubility could be increased by reducing the induction temperature [20]. Therefore, we firstly explored the optimum induction temperature while fixing the IPTG concentration and induction time, then the optimum IPTG concentration and lastly the optimum induction time. Since too low temperature condition will not facilitate the growth of cell, protein expression was induced at temperature 20○C in beginning. Additionally, high temperature and large number of exogenous protein can lead to the formation of inclusion body, which are considered as the major barrier to purify the production of target proteins [21]. So our highest induction temperature was 38○C. Finally, our results were consistent with other’s findings, and 20○C was the optimum temperature. With a lower induction temperature, the synthesis of target protein can be slowed down such that the target protein has sufficient time to fold correctly into a tertiary structure and where disulfide bonds can be properly matched, resulting in increased solubility [22].
After determining the optimal inducing temperature was 20○C for maximal solubility of expressed target protein, we sought to determine the optimal IPTG inducing concentration of transformed BL21 E. coli cultured with different IPTG concentrations for 4 h at 20○C. Our experiment indicated that the expression level was correlation to IPTG concentration, yet the overall trend of solubility was at opposite direction. Previous reports for various recombinant proteins have indicated that the higher IPTG concentration, the lower yield of soluble target protein and productivity [23-26]. High concentration of IPTG had been reported to reduce the growth rate of E.coli, particularly when the host bacteria express some toxic proteins or proteases, thereby reducing target proteins yields. The decreased growth rate of E.coli after induction with 6.25 1x10-2 mM of IPTG may be due to the negative effect of IPTG or the increasing burden of bacterial metabolism as a result of heterologous gene expression or due to protein toxicity [27-31].
Transformed BL21 cells were cultured with optimized IPTG induced concentration (1x10-4 mM) at 20○C for alternative time periods ranging from 2h to 10h. There was no significant correlation between solubility and induction time.
Conclusion
The research reported here shows that the level of GST-PTEN fusion protein expression is not significantly affected by induction temperatures ranging from 20○C to 38○C. However, expression of GST-PTEN protein increases with both increasing concentrations of IPTG and with increasing induction time period. In addition, the solubility of GST-PTEN protein decreased with both the increasing temperature and concentration of IPTG. We have demonstrated that the optimal strategy for expressing the highest soluble GST-PTEN protein yields was to induce E.coli at the exponential phase (OD 0.6-0.8) with 0.1 μM IPTG for 8 h at 20○C. Our research has identified optimal conditions needed to obtain a high yield of the soluble GST-PTEN fusion protein, and lay a foundation to further explore the functions of PTEN in vitro.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
ACKNOWLEDGEMENTs
This work is supported by the National Natural Science Foundation of China (No.31371386) and the Program for Excellent Talents in Henan Province (No.124200510010).
References
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]

