All published articles of this journal are available on ScienceDirect.

Phosphorylation on TRPV4 Serine 824 Regulates Interaction with STIM1
Authors Info & Affiliations
Abstract
The TRPV4 cation channel, a member of the TRP vanilloid subfamily, is expressed in a broad range of tissues where it participates in the generation of a Ca2+ signal and/or depolarization of membrane potential. Here, we identified stromal interaction molecule 1 precursor (STIM1) as an auxiliary protein of this epithelial Ca2+channel using confocal microscopy analysis and GST pull-down assay. The STIM1 protein associates specifically with the C-terminal tail of TRPV4 to form a complex. In previous reports, we demonstrated that the serine824 residue of TRPV4 is one of the target phosphorylation sites of serum/glucocorticoid regulated kinase 1 (SGK1). In this report we further identified the role of serine 824 phosphorylation. The TRPV4 mutant S824D (not S824A) exhibited a diminished capacity to bind STIM1. Using GST pull-down and co-immunoprecipitation assays, we demonstrated that STIM1 is part of the TRPV4 protein complex. Our observations clearly suggest that the formation of a complex between TRPV4 and STIM1 and its plasma membrane localization are regulated through phosphorylation of serine824 of TRPV4, and that the STIM1-TRPV4 complex plays crucial roles in routing TRPV4 to the plasma membrane from the endoplasmic reticulum and in maintaining its function.
INTRODUCTION
The TRPV4 cation channel, a member of the transient receptor potential (TRP) vanilloid subfamily, is expressed in a broad range of tissues where it participates in the generation of a Ca2+ signal and/or depolarization of membrane potential [1-4]. The participation of TRPV4 in osmo- and mechano-transduction contributes to important functions including cellular and systemic volume homeostasis, arterial dilation, nociception, epithelial hydroelectrolyte transport, bladder voiding, and ciliary beat frequency regulation [5-10]. TRPV4 also responds to temperature, endogenous arachidonic acid metabolites, and phorbol esters including the inactive 4α-phorbol 12,13-didecanoate (4-αPDD), and participates in receptor-operated Ca2+ entry, thus showing multiple modes of activation [11, 12]. In this regard, several proteins have been proposed to modulate TRPV4 subcellular localization and/or function including microtubule-associated protein 7, calmodulin, with no lysine protein kinases, and PACSIN3 [13-16]. In addition, a close functional and physical interaction exists between the inositol triphosphate receptor 3 (IP3R3) and TRPV4 that sensitizes the latter to the mechano- and osmo-transducing messenger 5'-6'-epoxieicosatrienoic acid (EET) [17, 18].
STIM1 is a 685-amino acid type I transmembrane protein that was originally identified in the plasma membrane, where it is oriented with its N-terminal domain targeted to the extracellular space [19]. STIM1 was initially described as a protein that induces growth arrest and degeneration of several human tumor cell lines, and is also involved in stromal-hematopoietic cell interactions [20]. When the intraluminal Ca2+ concentration is reduced, STIM1 re-localizes within the ER membrane to punctae at ER-plasma membrane junctions, which facilitates its association with members of the Orai-1 and transient receptor potential cation (TRPC) families [21, 22]. STIM1 has been reported to be the sensor of Ca2+ accumulation in the ER that activates store-operated Ca2+ entry (SOCE), a mechanism for Ca2+ influx that is controlled by the filling state of the intracellular Ca2+ stores [23]. Through this process, STIM1 activates the SOC channels until the ER Ca2+ levels are replenished [24, 25]. STIM1 has been reported to interact directly with Orai1 through a region located in the cytoplasmic ERM domain that was identified by different research teams and named either SOAR (STIM1 Orai-activating region), OASF (Orai-activating small fragment), CAD (CRAC-activating domain), or CCb9 [26-29]. In addition, it has been demonstrated that STIM1 is able to gate TRPC1 by electrostatic interaction between two conserved, negatively-charged aspartates in TRPC1 (D639, D640) and positively-charged lysine residues in STIM1 (K684, K685) located in the C-terminal polybasic region, suggesting that STIM1 plays an important role in the trafficking of transmembrane proteins such as Orai-1 and TRPC1 [30].
A number of regulatory proteins have recently been described that modify the biophysical, pharmacological, and expression properties of TRPV4 ion channels and transporters through direct interaction. These newly identified associated proteins have facilitated the elucidation of important molecular pathways modulating transport activity. Thus, the aim of the present study was to identify auxiliary proteins that interact specifically with the C-terminal domain of TRPV4. In the initial screen to identify proteins associated with TRPV4, STIM1 was proposed as a candidate protein, because it has been reported that the TRPC1-TRPV4 heterotetramer can bind the C-terminal tail of STIM1 [21]. Consequently we found that the protein-protein interaction between TRPV4 and STIM1 is regulated by phosphorylation of the serine 824 residue of TRPV4 [31, 32]. The functional interaction between TRPV4 and STIM1 was further substantiated by pull-down assays, immunehistologic studies, and Ca2+ ion image analysis. Thus, our data identify STIM1 as a new protein ligand that is involved in the regulation of TRPV4 subcellular localization and channel activity.
MATERIALS AND METHODS
Site-directed Mutagenesis
In order to obtain the mutants, amino acid changes were introduced using mutated oligonucleotides for S/A (up 5′-agg gat cgttggGccGcggtggtg ccc cgc gta-3′, down 5′-gcggggcacc accgCggCccaacgatccct acg-3′) or S/D (up 5′-agg gat cgttgg GAc GAC gtggtg ccc cgc gta-3′, down 5′-gcggggcaccac GTC gTCccaacgatccct acg-3′) and wild-type TRPV4 as a template. The TRPV4 mutant constructs were prepared using a QuickChange® Multi Site-Directed agenesis Kit (Stratagene). To obtain truncated TRPV4 (Δ718-871), we used primers (up 5′-ataggatccatgggt gag accgtgggc cag-3′, down 5′-atactc gag cta cag tggggcatcgtc cgt-3′). All TRPV4 mutants were confirmed via DNA sequencing. Human embryonic kidney (HEK293) cells were transfected with TRPV4 and with mutant constructs, as described previously.
Glutathione S-transferase (GST)-TRPV4 Fusion Proteins and Pull-down Assays
TRPV4 sequences were PCR-amplified, subcloned into pGEX-5X-1, sequenced, and expressed inEscherichia coli BL21. S824A-agarose or GST-TRPV4 fusion proteins bound to glutathione-Sepharose were equilibrated in PBS buffer containing 0.1% Triton X-100 and 1 mM CaCl2 or 2 mM EGTA. Incubation with the total cell lysates, S824A, or S824D was followed by three washes with the appropriate buffers, and the bound proteins were eluted with sample buffer, subjected to SDS gel-electrophoresis, and exposed to X-ray film (Fuji Las 3000 mini).
Fluorescence Measurements of [Ca2+]i
We measured [Ca2+]i using a fluorescent Ca2+ indicator Fluo4-acetoxymethyl ester (Fluo4-AM), as previously described. In brief, cells growing on coverslips were incubated for 40 min in DMSO solution containing 1 μM Fluo4-AM at 24 °C in darkness, and then washed and incubated for 15 min to hydrolyze internalized Fluo4-AM. We measured [Ca2+]i in single cells that emitted fluorescence, using confocal microscopy (LSM710 Zeiss, Germany) at wavelengths of 495 nm (excitation), and 519 nm (emission). The absorption (as an arbitrary unit) at 488 nm with an argon-ion laser was measured as a relative intracellular Ca2+ ion concentration [Ca2+]i. All experiments were carried out at 24 °C. After stimulation with mild heat (from 24 to 42 °C within 45 s for 2 min), [Ca2+]i was measured in single cells at a 24 °C solution.
CONFOCAL MICROSCOPY
MDCK or HEK293 cells were seeded overnight at 60% confluence onto culture slides coated with human fibronectin (Becton Dickinson, MA). The cells were washed several times with ice-cold PBS and fixed in 3% paraformaldehyde for 10 minutes. The fixed cells were permeabilized with 0.1% Triton X-100 for 10 minutes and blocked for 1 hour in PBS containing 5% BSA (Sigma, USA) and 0.1% Tween. Following incubation with a polyclonal antibody against STIM1 or monoclonal antibody against TRPV4, the cells were washed and stained further with a conjugated donkey anti-rabbit IgG prior to processing the slides for immunofluorescence. After an additional 20 minutes of incubation at 37 °C, the cells were fixed, permeabilized, and decorated with either an anti- STIM1 or TRPV4 antibody. As a secondary antibody, Alexa Fluor 568-conjugated donkey anti-rabbit or Fluor 488-conjugated goat anti-mouse (Molecular Probes, Inc., Eugene, OR) was used. Confocal microscopy analysis was performed LSM710 (Zeiss, Germany) at the Center for Experimental Research Facilities of Chungbuk National University.
SOLUTIONS AND DRUGS
Cells were superfused normally with a solution containing (mM): 88 NaCl, 5 KCl, 5.5 glucose, 1 CaCl2, 10 HEPES and 100 mannitol, adjusted to pH 7.4 with NaOH (300 mosm kg−1 H2O). The HTS was adjusted to 200 mosm Kg−1 H2O by omitting mannitol. 4-αPDD was acquired from Sigma (St Louis, MO, USA). Fluo-4AM and Fura-2AM were acquired from Molecular Probes, Inc (Eugene, OR, UK). Stock solutions of phorbol esters were initially prepared in dimethyl sulphoxide (DMSO) at a concentration of 1 mM, and then stored at –20 °C. The final DMSO concentration in the experimental bath solution containing phorbol esters never exceeded 0.5%. Insulin and SGK1 inhibitors GSK 650394 were acquired from Tocris Bioscience (Ellisville, MO, USA), and used them as the manufacturer's recommendation.
RESULTS
The Protein-protein Interaction between TRPV4 and STIM1 in MDCK Cell
STIM1 was initially identified as a TRPC1 or Orai1 interacting protein and the binding between both proteins was subsequently confirmed by GST pull-down assays [21, 30]. STIM1 is predominantly present as a heterotetrameric complex with Orai1 or TRPC1, which has been implicated in numerous biological processes including endocytosis, exocytosis and membrane-cytoskeleton interactions [21, 25, 30]. Thus, we assumed that the binding was not restricted to TRPC1, since TRPV4 was also shown to bind STIM1, indicating a mutual mechanism in the regulation of these Ca2+ channels. Considering the high degree of homology and the similarities in electrophysiological behavior between both channels, it is indeed likely that STIM1 and TRPV4 have common regulatory factors such as associated regulatory proteins (Fig. 1A). Recently, we identified that TRPV4 is also one of SGK1 substrate proteins (Fig. 1B), and the phosphorylation on serine 824 by SGK1 regulates the binding affinity to actin or tubulin [31]. To confirm whether endogenous TRPV4 interacts with STIM1 in the cell, we investigated whether the endogenous TRPV4 formed a protein complex with STIM1 in MDCK1 cells directly. As shown in Fig. (1c), the TRPV4 immunoprecipitate contained STIM1. Antibodies directed against STIM1 were also able to successfully capture TRPV4 from the same lysates, supporting the hypothesis that the two proteins are physically associated.
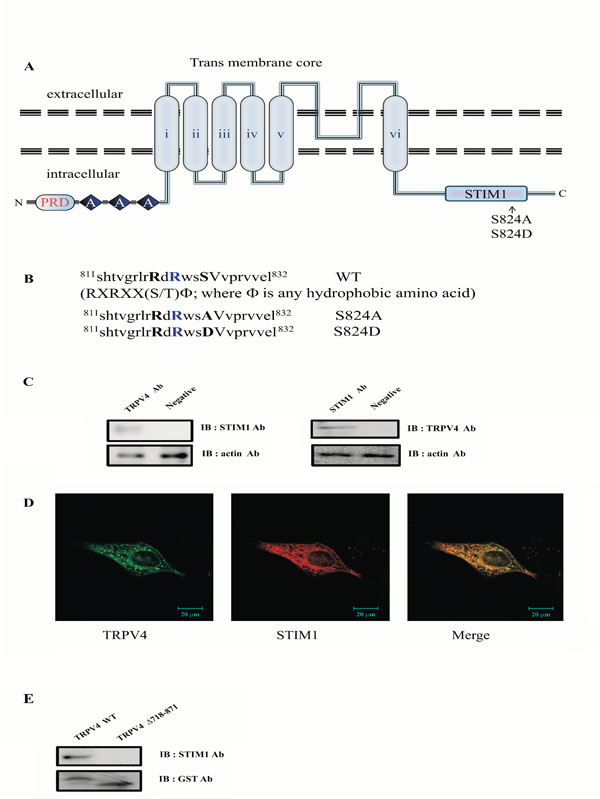
Protein-protein interaction between STIM1 and the C-terminal domain of TRPV4. (A) Transmembrane topology of mouse TRPV4 (871 aa). The three ankyrin binding repeats (ANK; gray circles), the six trans-membrane regions (TM1_TM6), the STIM1 binding site (STIM1), and the putative SGK1 phosphorylation site (serine824) are indicated. The C-terminal cytoplasmic region of TRPV4(Δ718-871) is also indicated. The putative SGK1 phosphorylation site (serine 824) is present in the STIM1 domain (812-832 aa) of the TRPV4 channel. The line indicates the GST C-terminal cytoplasmic region of TRPV4(Δ718-871) or mutant (S824A) fusion protein, the TRPV4 mutant site (S824A or S824D) is compared with the wild-type (WT; Gene Bank no. BC127052). (B) Alignment of TRPV4 WT, S824A, and S824D with the consensus SGK1 substrate motif. The putative SGK1 phosphorylation site (serine 824) of TRPV4 is located in the specific conserved SGK1 substrate [R-X-R-X-X-(S/T)ϕ]. S824A and S824D were constructed using site-directed mutagenesis. (C) Interaction between TRPV4 and STIM1 in MDCK cells. Following immunoprecipitation (IP) using an anti-TRPV4 antibody, immunoblot (IB) analysis was performed using an antibody against STIM1 (left). Conversely, STIM1 immunoprecipitated complexes were subjected to immunoblot analysis using an anti-TRPV4 antibody (right). Co-immunoprecipitation of STIM1 with TRPV4 confirmed the formation of a TRPV4-STIM1 complex. The negative control for immunoprecipitation was an unrelated antibody. The control for western blot analysis was an antibody against actin (bottom). (D) MDCK cells were examined by direct immunofluorescence microscopy. The figures show TRPV4 (green), STIM1 (red), and merged (yellow) confocal microscopic images. (E) The C-terminal of TRPV4 is required for interaction with STIM1. After incubation of GST-TRPV4WT or the deletion mutant fusion protein (lacking aa 718-871) with MDCK cell lysate, the purified GST bead was immunoblotted with an antibody against STIM1 (upper lane). The deletion mutant lacking the C-terminal (aa 718-871) did not pull down STIM1 (right lane), whereas GST TRPV4 WT did (left lane).
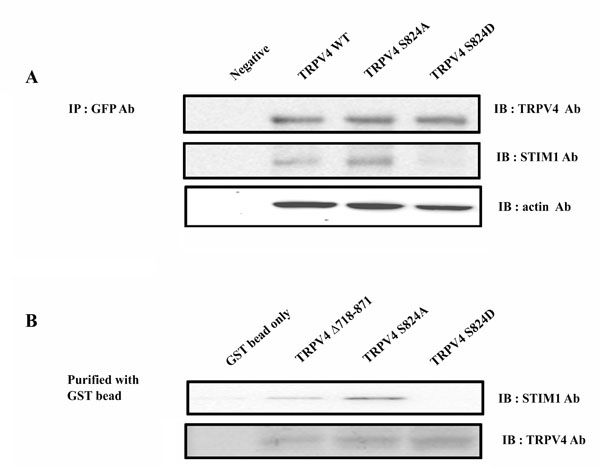
The effect of phosphorylation of TRPV4 serine 824 on the interaction between TRPV4 and STIM1. (A) Co-immunoprecipitation of TRPV4 WT, S824A, or S824D with STIM1. HEK 293 cells were transiently transfected with EGFP-TRPV4 WT or S824A plasmid. After 48 h the cells were lysated, and pull-down of total protein was conducted with protein Aagarose beads. Western blot assays were performed with rabbit anti-TRPV4 or anti-STIM1 antibody. (B) Pull-down analysis of STIM1 with GST fusion C-terminal TRPV4 WT, S824A, or S824D. GST-fusion proteins encompassing C-terminal TRPV4 domains were constructed and expressed in E. coli. Approximately 2 mg of WT, S824A or S824D fusion protein bound to the glutathione-Sepharose bead were incubated with HEK 293 cell lysates.
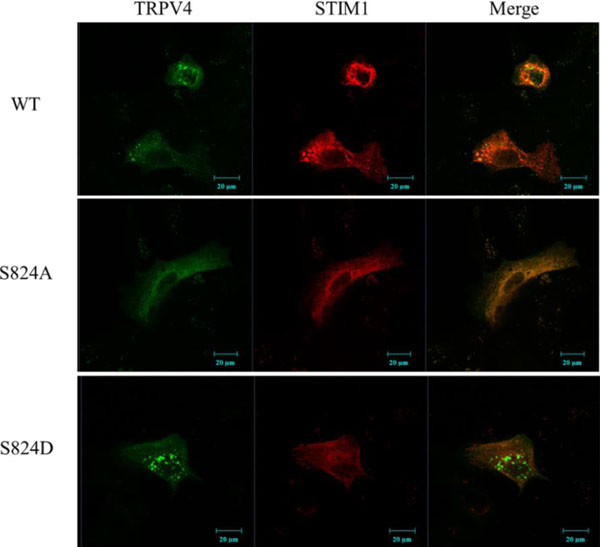
Confocal microscopic images of cells transfected with EGFP-TRPV4 WT, S824A,or S824D. Cells were examined by direct immunofluorescence microscopy. The figures show EGFP-TRPV4 WT or mutant (S824A or S824D) (green), pmCherry-N1 STIM1 (red), and merged (yellow) confocal microscopic images. EGFP-TRPV4 WT or S824A showed maximal colocalization with pmCherry-N1 STIM1 in the cytosol (upper and middle lane). However, EGFP-TRPV4 S824D was principally detected in the focal adhesions, and was minimal colocalized with STIM1 at the Golgi apparatus (down lane).
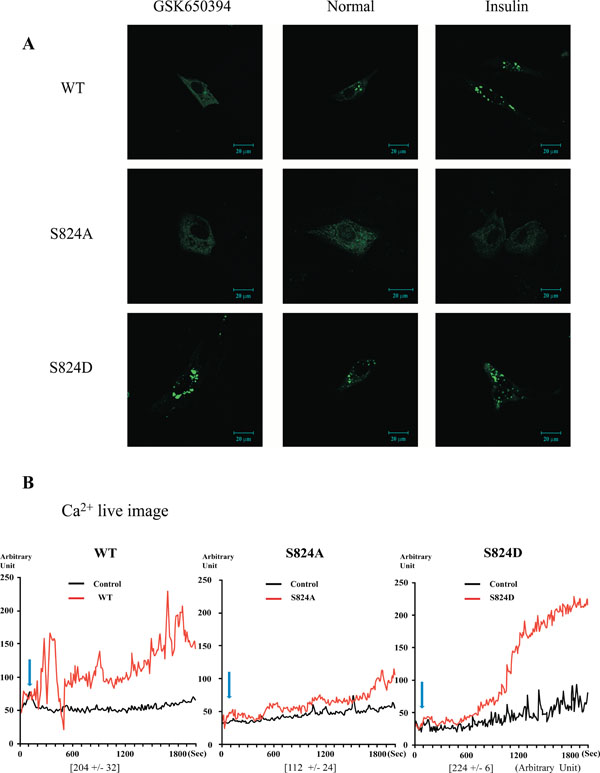
Plasma membrane localization of TRPV4 WT, S824A, or S824D in cells after treatment with GSK650394 or insulin. (A) Confocal microscopic analysis of the transfected EGFP-TRPV4 S824A, WT, or S824D (green) after treatment with SGK1 inhibitor (GSK 650397, left), activator (insulin, right) for 12 h, and the serum free control (middle). The localization of TRPV4 WT (which can be phosphorylated on serine824 by SGK1) was altered by treatment with GSK650394 or insulin. The image is representative of five repeat experiments. (B) Effects of 4-αPDD on intracellular calcium concentration change [Ca2+]i of TRPV4 WT, S824A, or S824D, expressed by the absorption at 488 nm of argon-ion laser in HEK 293 cells (as an arbitrary % unit).The average the maximum calcium flux represents below the graph (n=5).
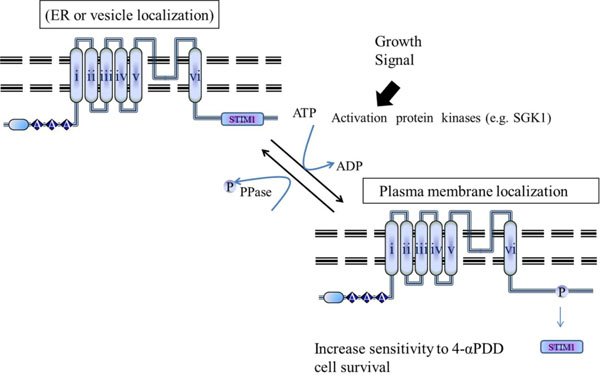
Putative model for the regulation of the interaction between TRPV4 and STIM1 by phosphorylation on TRPV4 serine 824. TRPV4 WT can be activated by the interaction with proteins such as STIM1 through association/dissociation from its C-terminal cytoplasmic domain. The mechanism determining density of TRPV4 in the plasma membrane seems to be similar to that of other channels such as GLUT4 or AQP2. In contrast to TRPV4 S824A, S824D mutant cannot associate with STIM1 (for more detail, see the Discussion section).
To determine whether TRPV4 exists together with STIM1 visually in MDCK cells, a confocal microscopy experiment was also performed. The results indicated that endogenous TRPV4 (green) and STIM1 (red) are co-localized in the cytosol (yellow) (Fig. 1D). These results also supported that the endogenous TRPV4 interacts with STIM1 directly in MDCK1 cells.
To further establish the interaction between TRPV4 and STIM1, GST pull-down binding assays were performed (Fig. 1E). The endogenous STIM1-containing MDCK homogenate was incubated with GST-TRPV4 WT or GST-TRPV4(Δ718-871) fusion proteins immobilized on glutathione-Sepharose 4B beads. STIM1 bound specifically to the C-terminal tail of TRPV4, since no interaction was observed with GST-TRPV4(Δ718-871) (Fig. 1 Eright lane). Thus, the C-terminal tail of TRPV4 is required for the interaction with STM1. Altogether, our observations suggested that TRPV4 interacts with STM1 through the C-terminal domain of TRPV4 in MDCK cell, directly.
Phosphorylation on TRPV4 Serine 824 Inhibits the Interaction between TRPV4 and STIM1
Previously we demonstrated that the interaction of TRPV4 with actin (or tubulin) is differentially regulated by the phosphorylation of TRPV4 serine 824 [31]. Similarly, we hypothesized that the phosphorylation of TRPV4 serine 824 also regulates the interaction between TRPV4 and STIM1. To demonstrate the requirement of TRPV4 serine 824 phosphorylation for the interaction between TRPV4 and STIM1, co-immunoprecipitations and GST pull-down assays of these proteins in their native form were performed (Fig. 2). HEK 293 cells which did not express their endogenous TRPV4 were transiently transfected with EGFP-TRPV4 WT or S824A plasmid. After 48 h the cells were lysated, and immunoprecipitation was conducted with mouse anti-TRPV4 or anti-STIM1 antibody. Western blot assays were performed with rabbit anti-TRPV4 or anti-STIM1 antibody (Fig. 2A). Interestingly, TRPV4 S824A (an analog the unphosphorylated TRPV4) brought down STIM1 (Fig. 2A middle lane), while TRPV4 S824D (an analog thephosphorylated TRPV4) and WT did not. Furthermore, STIM1 was more immunoprecipated by TRPV4 S824A than TRPV4 WT. These results suggested that the unphosphorylated TRPV4 (S824A) contained more affinity to STIM1 than the phosphorylated (S824D).
To confirm the hypothesis that the interaction between TRPV4 and STIM1 is regulated by the phosphorylation on TRPV4 serine 824 again, we performed the pull-down analysis of STIM1 with GST fusion C-terminal TRPV4 WT, S824A, or S824D (Fig. 2B). GST-fusion proteins encompassing C-terminal TRPV4 domains were constructed and expressed in E. coli. Approximately 2 mg of WT, S824A or S824D fusion protein bound to the glutathione-Sepharose bead were incubated with HEK 293 cell lysates. Similarly in Fig. (2a) result, STIM1 was precipitated from homogenates of HEK 293 cell using GST-TRPV4 C-terminal tail-loaded glutathione-Sepharose 4B beads, demonstrating a physical interaction between TRPV4 and STIM1 (Fig. 2B). Further, GST TRPV4 S824A brought down STIM1, while GST TRPV4(Δ718-871) and S824D did not. STIM1was more pull downed by TRPV4 S824A than TRPV4 WT, consistent with Fig (2A). Therefore, the observation supported that the phosphorylation on TRPV4 serine 824 inhibits the binding with STIM1.
The Requirement of Unphosphoryaltion on Ser 824 Residue of TRPV4 for Its Association with STIM1
STIM1 is predominantly present as a heterotetrameric complex with TRPC1, which has been implicated in numerous biological processes including endocytosis, exocytosis and membrane-cytoskeleton interactions. STIM1 is postulated to bind to the cytoplasmic face of membrane rafts to stabilize these domains, thereby providing a link to the actin cytoskeleton [21, 25].
To examine visually again whether the phosphorylation on TRPV4 serine 824 inhibits the binding with STIM1, HEK 293 cells were transiently transfected with EGFP-TRPV4 WT or S824A or S824D with pmCherry-N1 (Clontech, USA) STIM1 plasmid. After 48 h the cells were fixed, a confocal microscopy experiment was performed. The results indicated that exogenous TRPV4 (green) and STIM1 (red) are co-localized in the cytosol (yellow) (Fig. 3). As shown in Fig. (3) (upper lane), we observed that the exogenous TRPV4 WT interacts with STIM1 directly in HEK293 cells. Further, TRPV4 S824D was not co-localized with STIM1 (down lane), whereas TRPV4 S824A was co localized with STIM1 (middle lane), consistent with Fig. (2). Thus, these observations also supported our notion that the phosphorylation on TRPV4 serine 824 regulates not only the interaction between TRPV4 and STIM1 but also TRPV4 subcellular localization.
The Regulation of TRPV4 Activity and the Interaction with STIM1 by the Phosphorylation on TRPV4 Serine 824
Previously, we found that C-terminal domain of TRPV4 interacts with actin or tubulin, depending on the phosphorylation on TRPV4 serine 824 residue [31, 32]. Because STIM1 is also one of candidate proteins which interact with TRPV4 C-terminal domain, we predicted that the interaction with STIM1 is regulated by the phosphorylation on serine 824 residue of TRPV4. To determine whether the localization of TRPV4 and its interaction with STIM1 are changed by the phosphorylationon on TRPV4 serine 824, we tested the TRPV4 localization with a treatment of GSK650394 (SGK1 kinase inhibitor) or insulin (SGK1 activator). As control, TRPV4 S824A or S824D mutant was used (Fig. 4A). The subcellular localization of TRPV4 mutants was not changed by the treatment of GSK650394 or insulin, while WT was dramatically changed depending on the treatment.
The effect of STIM1 on TRPV4 activity was also determined by whole-cell Ca2+ image analysis in transiently transfected HEK293 cells. As shown in Fig. (4b), the S824A mutant of TRPV4 failed to produce significant Ca2+ currents in HEK293, while the channel activity of TRPV4 WT or S824D was readily detectable (Fig. 4B). Thus, these findings suggested that both the binding C-terminal of TRPV4 with STIM1 and TRPV4 subcellular (plasma membrane or focal adhesion) localization are regulated by the phosphorylation on TRPV4 serine 824 residue.
The Putative role of TRPV4 Serine 824 Residue Phosphorylation on its Ca2+-channel Activity Regulation
The present study demonstrated the presence of STIM1 in the TRPV4 channel complex, indicating a possible function in regulating channel localization and/or activity. Previously we demonstrated that the serine 824 residue of TRPV4 is one of SGK1 phosphorylation sites (Figs. 1-4). Therefore, it is proposed that the phosphorylation on the residue reduces its protein-protein interaction with STIM1, directly, resulting in the enhancement of TRPV4 density in the plasma membrane (Fig. 5).
In conclusion, we provided the first evidence of a regulatory role for the STIM1-TRPV4 complex in Ca2+ (re)absorption in general and in particular in TRPV4 routing. The interaction between STIM1 and TRPV4 was regulated by the phosphorylation on TRPV4 serine 824 residue. The elucidated molecular mechanism involves tethering of the STIM1-TRPV4 complex to the Ca2+ channel, resulting in functional plasma membrane localization. This mechanism is likely applicable to other ion transporters given the wide tissue distribution of TRPV4 and the associated STIM1 protein. The phosphorylation on TRPV4 serine 824 residue seems to be the major regulatory point for its plasma membrane localization and the interaction with other proteins including STIM1, IP3R3, calmodulin, actin, or tubulin [13-17, 31].
DISCUSSION
The present study identified the TRPV4-STIM1 pair as the first auxiliary protein complex for the epithelial Ca2+ channels, TRPV4 (Fig. 1). STIM1 forms a well-defined heteromeric complex with Orai1 and TRPC1 [20, 21]. Our data also provided the first evidence of a regulatory role for the STIM1-TRPV4 complex in Ca2+ (re)absorption in general. The interaction between STIM1 and TRPV4 was also regulated by the phosphorylation on TRPV4 serine 824 residue (Fig. 1B). Thus, the phosphorylation on TRPV4 serine 824 residue is suggested as the major regulatory point for the interaction with other proteins including STIM1, calmodulin, IP3R3, actin, or tubulin (Fig. 5) [13-16]. STIM1 was originally identified in a screen for stromal cell gene products that bound to preB cells [19]. This gene is conserved in sequence from Drosophila to mammalian species and appears to be expressed ubiquitously in human tissues, based on published analyses of mRNA and protein expression [22]. Thus, the interaction between TRPV4 and STIM1, which negatively regulate TRPV4 channel activity or its plasma membrane channel density through the sequestering TRPV4 around ER (Figs. 3 and 5), may contribute to elucidate the protein functions.
TRPV4 channels in vivo are often composed of heteromeric subunits [1, 21, 32]. Heteromeric assembly usually occurs between the members within the same TRP subfamily, such as between TRPC1 and TRPC4 [21]. However, co-assembly could also happen between the subunits from a different TRP subfamily, such as between TRPC1 and polycystic transient receptor potential 2 (TRPP2) (PKD2 and between TRPV4 and TRPP2 [21, 33]. These heteromultimeric channels may display properties different from those of homomultimeric channels [1, 21, 32, 33]. To our knowledge, there is still no report about the vesicular translocation of TRPV4 channels. Recent others indicated that TRPC1 can co-assemble with TRPV4 to form heteromeric channels [32, 34]. In vascular tissues, the TRPV4-C1 heteromeric channels are the main channels responsible for flow-induced endothelial Ca2+ influx and subsequent vascular relaxation [34]. In separate studies, it was reported that SGK1 phosphorylation potentiates flow-induced Ca2+ influx and subsequent its plasma membrane localization [4, 31]. Therefore, we propose the hypothesis that the SGK1 phosphorylation may enhance TRPV4 channel density in the plasma membrane through the dissociation from STIM1, similar with the regulation mechanism of GLUT4 or AQP2 by insulin or vasopressin, respectively (Fig. 1B, and Fig. 5) [35, 36]. However, the existence of vesicle containing TRPV4 in the cell remains to be proven.
The high Ca2+-selectivity of Orai1 channels in the presence of extracellular Ca2+ has been suggested that STIM1 located in the plasma membrane modulates store-operated divalent cation entry by interaction with TRPC1 channels. Even though the functional properties of plasma membrane-resident STIM1 remain largely uncharacterized, the function of ER-located STIM1 has been widely accepted as the adapt protein to attach the partner protein at ER. Because TRPV4 S824A contains more affinity to STIM1 than S824D and WT, S824A is shown more in the cytoplasm, not in the focal adhesion spots of plasma membrane (Fig. 3).
Ma et al. reported that TRPV4-C1 heteromeric channels were more favorably translocated to the plasma membrane than TRPC1 or TRPV4 homomeric channels, and formed a protein complex with STIM1 [21]. Further, they suggested thatthe enhanced TRPV4-C1 insertion to the plasma membrane could contribute to SOCE and SOC [21, 24, 25]. They also suggested that Ca2+ store depletion enhances trafficking of heteromeric TRPV4-C1 channels to the plasma membrane. Because of the existence of heteromeric TRPV4-C1, we cannot rule out the possibility that the interaction between STIM1 and TRPV4 is mediated by other proteins, such as TRPC1. In our experiment here, we used the purified TRPV4 fusion protein to exclude the endogenous TRPC1 interaction with TRPV4 (Fig. 2). However, we do not rule out the possibility of intermediating proteins in the interaction between TRPV4 and STIM1, such as TRPC1.
Other researchers reported that STIM1 binds TRPCs via its electrostatic charge, the association mediated by the electrostatic charge has been found to be sufficient to activate the channels [29]. However, our results indicate that TRPV4 mutation (823DD824→ 823AA824) did not impairs the sensitivity to extracellular Ca2+ of divalent cation entry operated by the intracellular Ca2+ stores. Furthermore, the STIM1 (685KK686→ 685AA686) mutation did not prevent STIM1-TRPV4 interaction (data not shown). Thus, it seems to be that STIM1 binds with TRPV4 by its protein-protein interaction, but not by its electrostatic charge.
Previously, we have already demonstrated that TRPV4 channel is one of SGK1 authentic substrate proteins, and that the serine 824 residue of TRPV4 is phosphorylated by SGK1 (Fig. 1B) [31, 32]. And we demonstrated that phosphorylation on the serine 824 residue of TRPV4 is required for its interaction with F-actin, using TRPV4 mutants (S824D and S824A) and its proper subcellular localization [31, 32]. In the line of expansion, we showed here that the phosphorylation on TRPV4 serine 824 residue promotes its dissociation from STIM1 for its plasma membrane localization (Fig. 2). The signal convergence on the C-terminal domain of TRPV4 may show an important mechanism by which the timing and convergence of signal responses is integrated [2]. Our results provide evidence for a functional role of plasma membrane-resident TRPV4 in the regulation of store-operated Ca2+ entry by its C-terminal domain through the protein-protein interaction (Fig. 1A).
Recently, we also reported that the binding of Ca2+-calmodulin (CaM) can be prevented by the SGK1-mediated phosphorylation on the serine 824 residue within the CaM binding site [31, 32]. Conversely, the substitution of the target site (serine 824) with aspartic acid (S824D) results in a more rapid and longer activation of TRPV4-mediated currents, as Ca2+-CaM binding can be inhibited by phosphorylation, though the negative feedback regulator at high Ca2+ concentration [2, 4]. Even though it was demonstrated that the phosphorylation on TRPV4 serine 824 occurs by SGK1 (Fig. 1B), the serine can also be phosphorylated by other protein kinases, such as Akt kinase and protein kinase A. The elucidation how the outside signal relays to TRPV4 activation is required to better understand TRPV4 regulation mechanism (Fig. 5). TRPV4 plays an important role in pathological sensory perception and bone growth. Because the potential effect of a TRPV4 functional mutation in human genetic disease remains to be elucidated, the characterization TRPV4 interacting protein may be also useful to cure or alleviate the human disease caused by TRPV4 mutations [1-5].
CaM is characterized a ubiquitous dual function protein, which is known to regulate the activity of different ion channels, Ca2+ pumps and other proteins in a Ca2+-dependent manner [2]. Similarly, we assumed that the association of STIM1 with TRPV4 inhibits TRPV4 channel activity in the plasma membrane. The activation of TRPV4 S824D seems to be achieved via preventing an inhibitory protein (such as Ca2+-CaM and STIM1) association (Fig. 4B).
TRPV4 contains a consensus sequence for the phosphorylation by SGK1 within the CaM binding domain (aa 811-850) (Fig. 1B). The subcellular localization of TRPV4-S824A or S824D was independent on the SGK1 modulator treatment (Fig. 4A), while that of TRPV4-WT was effected. Thus, both STIM1 and CaM are able to affect TRPV4 channel function through the protein-protein interaction. However, we do not know whether these two different proteins effect on TRPV4 function synergically or competitively. Because it is not clear that unidentified proteins also modulate TRPV4 function through the protein-protein interaction with its C-terminal domain, the characterization of the proteins interacting with the domain will be helpful to understand the role of C-terminal domain of TRPV4 [2].
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
This work was supported by Korea Research Foundation Grant (NRF-2013R1A1A4A01005522) to S. S. Kang. Both Shin and Lee are Korean Research fellowship recipients (NRF-2014R1A1A2009622, NRF-2013R1A1A2063994). We thank to Dr. David M. Cohen (Oregon Health and Science University) for TRPV4 gene, and to Dr. Joong-Kook Choi (Chungbuk National University) for pmCherry-N1 STIM1 construct.

