All published articles of this journal are available on ScienceDirect.

The CCA-end of P-tRNA Contacts Both the Human RPL36AL and the A-site Bound Translation Termination Factor eRF1 at the Peptidyl Transferase Center of the Human 80S Ribosome
Authors Info & Affiliations
Abstract
We have demonstrated previously that the E-site specific protein RPL36AL present in human ribosomes can be crosslinked with the CCA-end of a P-tRNA in situ. Here we report the following: (i) We modeled RPL36AL into the structure of the archaeal ortholog RPL44E extracted from the known X-ray structure of the 50S subunit of Haloarcula marismortui. Superimposing the obtained RPL36AL structure with that of P/E tRNA observed in eukaryotic 80S ribosomes suggested that RPL36AL might in addition to its CCA neighbourhood interact with the inner site of the tRNA elbow similar to an interaction pattern known from tRNA•synthetase pairs. (ii) Accordingly, we detected that the isolated recombinant protein RPL36AL can form a tight binary complex with deacylated tRNA, and even tRNA fragments truncated at their CCA end showed a high affinity in the nanomolar range supporting a strong interaction outside the CCA end. (iii) We constructed programmed 80S complexes containing the termination factor eRF1 (stop codon UAA at the A-site) and a 2’,3’-dialdehyde tRNA (tRNAox) analog at the P-site. Surprisingly, we observed a crosslinked ternary complex containing the tRNA, eRF1 and RPL36AL crosslinked both to the aldehyde groups of tRNAox at the 2’- and 3’-positions of the ultimate A. We also demonstrated that, upon binding to the ribosomal A-site, eRF1 induces an alternative conformation of the ribosome and/or the tRNA, leading to a novel crosslink of tRNAox to another large-subunit ribosomal protein (namely L37) rather than to RPL36AL, both ribosomal proteins being labeled in a mutually exclusive fashion. Since the human 80S ribosome in complex with P-site bound tRNAox and A-site bound eRF1 corresponds to the post-termination state of the ribosome, the results represent the first biochemical evidence for the positioning of the CCA-arm of the P-tRNA in close proximity to both RPL36AL and eRF1 at the end of the translation process.
INTRODUCTION
It is general wisdom that peptide-bond formation is a function of the rRNA of the large ribosomal subunit (“the ribosome is a ribozyme” [1, 2]). Indeed, models for the mechanism of peptide bond formation on the ribosome use the structural insights from the crystallographic studies demonstrating that ribosomal proteins do not play a major role in the active site of the ribosome. For example, as reported by Nissen et al. [1], in the case of the archaeon Haloarcula marismortui (Hma), “the refined three-dimensional structure of a 50S ribosomal subunit at a resolution of 2.4 Å reveals a void of protein electron density in a radius of 18 Å around the peptidyl transferase center (PTC)”. In the case of eukaryotic ribosome, the few available 3-D structures reveal an increased PTC-complexity compared to its prokaryotic counterpart ” [3-5]. In fact, protein and rRNA components of eukaryotic ribosomes contain long eukaryote-specific extensions and several of the proteins are exclusively present in eukaryotes. Therefore, it is largely accepted that there might exist important differences between bacterial and eukaryotic ribosomes with regard to the arrangement of the eukaryote-specific extensions in the 3-D structures. Moreover, the ribosome is an inherently dynamic structure, and one can easily conceive that, with regard to the X-ray structures of the ribosome, biochemical approaches are more suitable to follow conformational changes in macromolecular complexes in solution, and will provide complementing information on the mutual arrangement of specific ribosomal components.
Interestingly, we have previously demonstrated the presence of the eukaryote-specific large subunit ribosomal protein L36AL (RPL36AL) near the PTC of the human 80S ribosome [6]. This protein was shown to contact the CCA-end of a deacylated tRNA at the P/E hybrid site on human 80S ribosome [6, 7], in agreement with crystallographic data on the large ribosomal subunit of the archaeon Hma [8]. We applied a zero-distance affinity labeling with periodate-oxidized tRNA (tRNAox). This approach was successfully used for mapping the active site of purified aminoacyl-tRNA synthetases thirty years ago [9, 10] leading to the discovery of the KMSKS motif that is considered today as a signature for the active site of class 1 aminoacyl-tRNA synthetases [11].
According to the X-ray data, the L44e protein, which is the archaeal counterpart of RPL36AL, is located at the large ribosomal subunit E-site, where it interacts with the two unstacked cytosine residues C75 and C74 of a native tRNA through an extension loop of the protein [8]. Since ribosomal proteins from the L44e family present an extended 3-D structure, we test here the hypothesis that caused by conformational changes of the ribosome during the tRNA shifts between classical and hybrid-site positions, the human E-site specific L36AL protein flips from the E- to the P-site neighbourhoods where it could interact with the P-tRNA. Here we present evidence that indeed the ribosomal protein L36AL interacts with a P-tRNA outside the CCA-end at the 3’-terminus. Furthermore, we demonstrate that the translation termination factor eRF1 bound to the A-site in the presence of a stop codon lies in close proximity to both the P-tRNA and the endogenous L36AL protein, at the PTC of human 80S ribosomes.
RESULTS
Modeling of the Human L36AL Protein and of Its Interaction with tRNA
We modeled the 3-D structure of human RPL36AL by homology with the homologous RPL44E of the 50S Hma ribosomal subunit (Fig. 1A), the X-ray crystal structure of which had been determined in a complex with a deacylated tRNA minihelix [8]. We have previously shown that the lysyl residue 53 could be crosslinked to cytidine75ox of tRNAAsp75ox [7], and we have described the post-translational modifications including monomethylated Gln-51 of the 49GGQ51 motif [7, 12] (Fig. 1A). Not surprisingly, the model 3-D structure of RPL36AL was close to that of canine RPL36A observed in a previous study by Chandramouli and co-workers [13]. In this 3-D structure, the methylated GGQ motif (green in Fig. 1A) of RPL36AL occupies a position comparable with that of canine RPL36A and with the corresponding GND motif of the archaeal counterpart L44E from Hma [8] in a loop at one extremity.

Model of the L36AL protein and of its interaction with tRNA. (A), ribbon representation of the 3-D structure of human RPL36AL (fragment 1-94) modeled by homology with the crystallographic structure of the archaeal counterpart RPL44E of the 60S ribosomal subunit from Haloarcula marismortui. The post-translational modifications (including the methylated Q51) [7], and the consensus pattern 61Kx(TorV)KKxxL(KorR)xxC72 (numbering of human RPL36AL) of the L44e family of r-proteins are colored pink and cyan, respectively. The 49GG50 dipeptide of the GGQ motif is highlighted in green. A zinc ion represented by a cadmium colored yellow is also shown. Fragment 86-94 corresponding to the nucleotide binding motif 2 (NBD2) common to all eukaryotic RPL7 [29] and RPL36A/RPL44 is shown in wheat. (B), overlaid structures of tRNAPhe (PDB ID 1JGQ) colored grey and of human RPL36AL (blue). The GGQ motif is shown in red, the 3’ terminal CCA trinucleotide of tRNA in pink, and the side chain of Lys-53 in green. (C), NBD2 [29] located in the C-terminal region of the eukaryotic RPL7 family (ortholog of the bacterial L30 protein) is conserved in the C-terminal region of the eukaryotic RPL36A/RPL44 family. The organisms are: Arabidopsis thaliana (arb. thal.), Drosophila melanogaster (droso.), Saccharomyces cerevisiae (s. cer.), Solanum tuberosum (sol. tub.). The bottom line labels residues as either strictly conserved (*), highly conserved (:) or weakly conserved (.). The alignment was generated with the program ClustalX.
Using the derived L36AL structure, we modeled the interactions between this protein and tRNA. Fig. (1B) shows the overlaid 3-D structures of tRNAPhe (PDB ID 1JGQ) and the modeled human RPL36AL, suggesting that the protein interacts with tRNA at the inner side of the elbow. This mode of interaction is reminiscent of the general model of tRNA-aminoacyl-tRNA synthetase interaction which shows, as described by Chu and Horowitz [14], that “synthetases bind their cognate tRNAs along and around the inside of L-shaped structure, recognizing the acceptor stem and, depending on the particular enzyme and tRNA, possibly also the D-stem and the anticodon”. The high-resolution crystal structure of the E. coli glutaminyl-tRNA synthetase-tRNAGln-ATP complex [15] is representative of this model, which is the source of the specificity and fidelity of the aminoacylation reaction.
Binding Assays on Biacore
The possible interactions between a P-tRNA and the protein L36AL, and the similarity to tRNA•synthetase interactions prompted us to test whether isolated recombinant L36AL can bind tRNA. We used the Biacore assay, which allows an easy assessment of the strength of a possible interaction. To this end, we used various tRNA fragments for the binding assays. In addition to the full length tRNA (tRNAAsp76), we tested tRNA species that were shortened by one (tRNAAsp75), two (tRNAAsp74), three (tRNAAsp73), four (tRNAAsp72) or five (tRNAAsp71) nucleotides from the 3’-end. To determine the binding affinities of RPL36AL to tRNA variants, we carried out surface plasmon resonance (SPR) analyses with a Biacore biosensor, which provides real-time protein:RNA interaction data by measuring the change in refractive index at the surface of the sensor chip due to the interactions (Figs. 2A and 2B). Experimental data from individual kinetic binding experiments were analyzed and fitted using BIAevaluation software with 1:1 binding model, in accordance to the 1:1 stoichiometries of binding of tRNA to the aminoacyl-tRNA synthetases polypeptide chains [9,16].
The resulting kinetic constants given in Table 1 revealed that the interactions between full length or truncated tRNA molecules and purified recombinant human RPL36AL take place with association (ka or kon in the range 0.76-1.55 X 10+6 M-1.s-1) and dissociation rates (kd or koff in the range 1.65-3.17 X 10-3 s-1), defining strong binding affinities (KD values in the range 1.23-3.02 X 10-9 M). The KD values in the nanomolar range reflect high tRNA-binding affinities with full length and truncated tRNA species. As a control, poly(A) or poly(U) RNAs of about one hundred nucleotides, as well as the combination of equimolar concentration of both, failed to bind to the protein (results not shown).
Kinetic and affinity constants for each tRNA species, including association (ka or kon) and dissociation (kd or koff) rates and affinity constants (KD), as deduced from the analyses in Fig. (2). The experimental errors on the KD were in the range 5-7% of the values reported in this Table
Kinetic and affinity constants for the interactions between a P-tRNA and the protein L36AL.
| tRNA species | ka = kon (x 10+6 M-1.s-1) | kd = koff (x 10-3 s-1) | KD = kd/ka (x 10-9 M) |
|---|---|---|---|
| tRNA Asp76 | 1.2 | 2.6 | 2.2 |
| tRNA Asp75 | 1.3 | 1.7 | 1.2 |
| tRNA Asp74 | 1.5 | 2.3 | 1.5 |
| tRNA Asp73 | 1.0 | 3.2 | 3.0 |
| tRNA Asp72 | 0.7 | 1.8 | 2.4 |
| tRNA Asp71 | 1.3 | 1.6 | 1.2 |
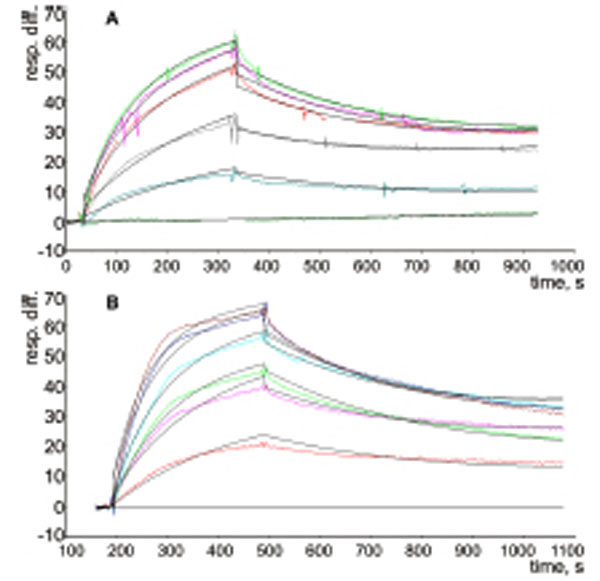
Kinetic measurement of RPL36AL:tRNA interaction. (A), His-tagged RPL36AL was immobilized on the surface of NTA sensor chip at low density resonance units (RU). Various concentrations of tRNAAsp76 (0, 1, 2, 4, 5 and 6 nM) were run over the chip surface. (B), the same experiment with tRNAAsp71.
Crosslinking of Human Recombinant RPL36AL with the Periodate-oxidized tRNAs
The full length tRNA molecule (tRNAAsp76), or the truncated tRNA species mentioned in the preceding section were treated with sodium periodate and the obtained 2’,3’-dialdehyde derivatives (tRNAox) were used for the crosslinking studies with isolated recombinant RPL36AL. Equimolar amounts of human recombinant RPL36AL were incubated in the presence of tRNAox (200 picomoles each), followed by an analysis of the crosslinks on urea-PAGE. Fig. (3) shows that a covalent complex is formed (band “a”), with a crosslinking yield of practically 100%, except in the cases of tRNA76ox and tRNA75ox (lanes 1 and 2, respectively). In fact, during the polyacrylamide gel electrophoresis in urea (Fig. 3), only tRNAox (lower band) and the tRNAox-L36AL covalent complex (upper “a” band) with their 75 (or so) negative charges of the phosphate groups would migrate toward the (+) pole at the bottom of the gel, while the free L36AL protein which is very basic (pI 10.59) would migrate toward the (-) pole at the top of the gel. As a consequence, the free L36AL would not even penetrate the urea-PAGE gel. Therefore, the non-crosslinked proteins in lanes 1 and 2 should not be visible. The possibility that the ribosomal protein could migrate as an unspecific, non-covalent complex together with the tRNA owing to the high binding affinity was ruled out by an SDS-PAGE analysis: the RPL36AL-tRNAox crosslinks showed RPL36AL-tRNAox covalent complexes corresponding to the band “a” in Fig. (3). Furthermore, the band “a” in Fig. (3) migrated with about 40,000 Da (revealed by comparison with standard marker proteins) indicating a 1:1 covalent complex between the His-tagged recombinant RPL36AL (MW of 15,000 Da) and the tRNAox species (MW of 25,000 Da, on average) (results not shown). Finally, the crosslinking yield with tRNA76ox is weaker than with the shortened tRNAox species (Fig. 3), in agreement with previously reported crosslinking data with the endogenous ribosomal protein L36AL in situ [7].
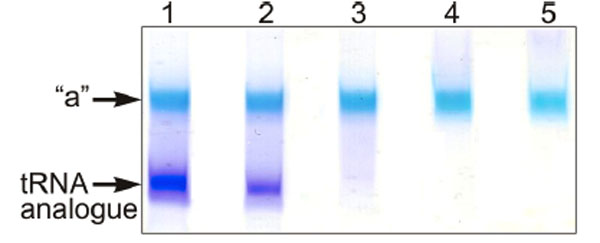
Crosslinking of recombinant human RPL36AL with periodate-oxidized full length tRNA (tRNAAsp76ox, lane 1), or tRNAox species that were shortened by one (tRNAAsp75ox, lane 2), two (tRNAAsp74ox, lane 3), three (tRNAAsp73ox, lane 4) or four nucleotides (tRNAAsp72ox, lane 5) from the 3’-end. The incubation mixtures were applied onto a 10% polyacrylamide gel run by 8 M Urea electrophoresis and colored with Stains all. The “a” bands represent the tRNAox species crosslinked with recombinant L36AL, while the lower bands represent the tRNA analogues, as verified by routinely performed control experiments on the full length tRNA molecule (tRNAAsp76), or the truncated tRNA species.
The CCA End of P-tRNA Contacts Both the Human Ribosomal Protein L36AL and the Translation Termination Factor eRF1 Bound to an A-site Displaying a Stop Codon
Similarly to Phe-tRNAPhe used previously, whose anticodon base-pairs with an A-site UUU triplet adjacent to the P-site bound tRNAox in an elongating ribosome [7], the termination factor eRF1 occupies the A-site in the presence of a stop codon during translation termination. What, if any, are the contacts between eRF1 and the deacylated P-tRNA at the end of the translation process? We constructed two complexes, both contained a tRNAAsp76ox at the P-site with a free A-site (complex 1) or with the eRF1 factor at the A-site (complex 2, see Fig. 4).
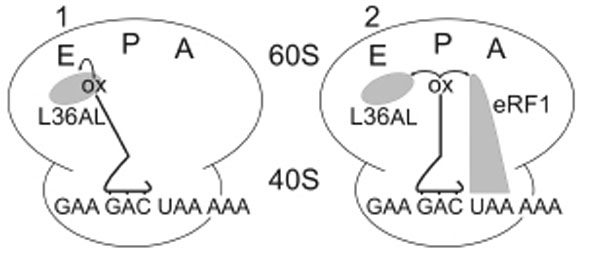
Complexes 1 and 2 designed for crosslinking of Human 80S ribosome with P-tRNAAsp76ox in the absence or in the presence of human eRF1 bound to an A-site UAA stop codon.
We tried formaldehyde and glutaraldehyde as crosslinking reagents, but failed to detect crosslinks between the [32P]-labeled P-tRNAox and eRF1 or any ribosomal component. Since formaldehyde is dedicated to the crosslinking of macromolecules that are in close contact, failure of this reagent would suggest that the P-tRNA is not in a close contact with either the L36AL protein or the eRF1 at the A-site. An alternative possibility is that the absence of a crosslink could reflect the absence of suitable targets for crosslinking in the contact regions of these macromolecules. Surprisingly, bands whose electrophoretic mobility corresponded to distinct covalent complexes made of L36AL, tRNAox and eRF1 were observed on the SDS-PAGE gel in the absence of reagents (Fig. 5A, lane 1). These findings suggested that the two aldehyde groups of tRNAox react with both L36AL and eRF1 (Fig. 6). Comparison of the migration of the crosslinked complexes “a” and “b” to that of protein markers on the same gels indicated apparent molecular weights of 36,000 + 1,000 Da (“a”) and 87,000 + 2,000 Da (“b”). The molecular weight of the band “a” agreed surprisingly well with that predicted for a binary complex containing one molecule of endogenous RPL36AL (12,000 Da) and one of tRNAox (25,000 Da; calculated MW of the binary complex 37,000 Da). Likewise, the MW of complex “b” agreed well with a ternary complex that contained in addition to the binary one one molecule of eRF1 (48,000 Da) leading to a calculated MW of 85,000 Da (ternary complex RPL36AL-tRNAox-eRF1 “b”, see Figs. 5A, 5C and 6). We note that, when the crosslinking of human 80S ribosomes was performed with an elongator tRNA-Met-ox positioned at the P-site in the presence of an appropriate mRNA and of eRF1 bound to an A-site stop codon, the ternary complex “b” was formed with comparable crosslinking yields (results not shown) suggesting that both tRNA dialdehyde derivatives, when bound to the P-site, behave similarly in the crosslinking reaction.
Also the shorter tRNA variants [5’-32P]tRNAAsp75ox, [5’-32P]tRNAAsp74ox or [5’-32P]tRNAAsp73ox were able to generate crosslinked complexes “a” and “b” (Fig. 5A, lanes 2-4, respectively). Since the crosslinked radioactive material is distributed between the binary RPL36AL-tRNAox (“a”) and the ternary RPL36AL-tRNAox-eRF1 (“b”) covalent complexes, the sum “a”+”b” represents the maximum of crosslinking (i.e. 100%) under a distinct experimental condition. The corresponding fractions of the “b” complex as percent of the total crosslinked radioactive material were estimated to 22%, 1.3%, 1.3% and 1.4% corresponding to lanes 1-4. There is a dramatic decrease of crosslinking yields of the ternary complex upon removal of A76 of the CCA-arm.
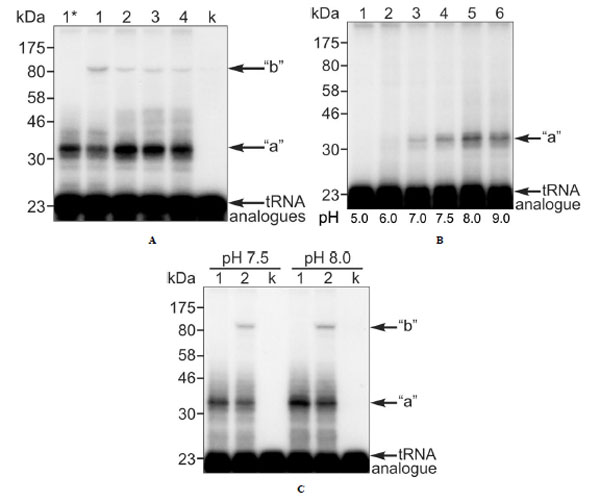
Crosslinking of endogenous ribosomal protein L36AL in human 80S ribosomes with full length or truncated tRNAox species in the absence or in the presence of the translation termination factor eRF1. (A) Complexes 2: 80S ribosomes incubated in the presence of eRF1 with [5'-32P]tRNAAsp76ox, [5'-32P] tRNAAsp75ox, [5'-32P]tRNAAsp74ox or [5'-32P]tRNAAsp73ox (lanes 1-4, respectively). The controls are: (i) crosslinking in ribosomal complex 1 with [5'-32P]tRNAAsp76ox, in the absence of eRF1 (lane 1*); (ii) crosslinking of [5'- 32P]tRNAAsp76ox with eRF1 in the absence of 80S ribosomes (lane k). (B) 80S ribosomes incubated with [5'-32P]tRNAAsp75ox at pH 5.0, 6.0, 7.0, 7.5, 8.0 and 9.0 (lanes 1-6, respectively) with a free adjacent A-site. Sodium borohydride (NaBH4) was used to trap the fraction of unprotonated reactive ε-amino group that is responsible for the nucleophilic addition to the aldehyde in order to form the Schiff base. (C) Same experiment as in (A) with 80S ribosomes carrying [5'-32P]tRNAAsp76ox at the P-site and eRF1 at the A-site incubated at pH 7.5 and 8.0, respectively (lanes 2) with the following controls. Lanes 1, crosslinking in ribosomal complex 1 with [5'-32P]tRNAAsp76ox, in the absence of eRF1 at pH 7.5 and 8.0, respectively. Lanes k, crosslinking of [5'-32P]tRNAAsp76ox with eRF1 in the absence of 80S ribosomes at pH 7.5 and 8.0, respectively (lanes k). For details see Materials and Methods.
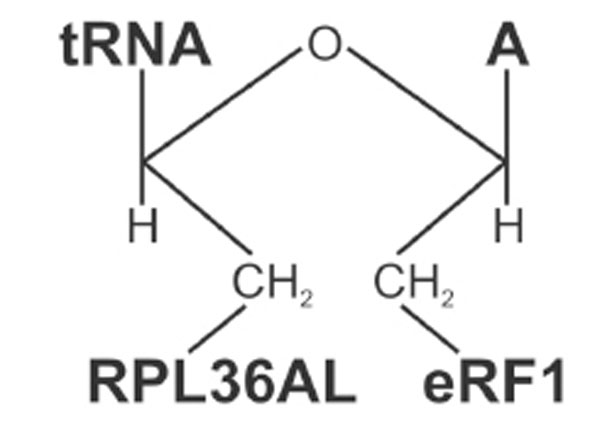
Structure of the ternary RPL36AL-tRNAox-eRF1 covalent complex.
We anticipated that the amount of ternary complex “b” would depend on that of the binary complex “a”. Therefore, we compared the yield of the binary RPL36AL-tRNAox complex “a”, using [5’-32P]tRNAAsp75ox and a pH of 8.0 used in a previous report, where we identified Lys-53 of this ribosomal protein as the tRNAox crosslinked site [7]. As shown in Fig. (5B), lane 5, a higher yield of crosslinked binary RPL36AL-tRNAox complex “a” was obtained at pH 8.0 in contrast to a pH 7.5 used in Fig. (5A).
Accordingly, we compared the efficiency of formation of the ternary crosslinked complex RPL36AL-[5’-32]P tRNAAsp76ox-eRF1 (complex “b”) at pH 7.5 (the usual crosslinking pH [7]) and pH 8.0. The relative yields of complex “b” were 22% at pH 7.5 and 34% at pH 8.0 (lanes 2 in Fig. 5C). In a further control we analyzed, whether after a crosslink between tRNAox and RPL36AL (in the presence of NaBH3CN that selectively reduces only the imminium group), the remaining aldehyde group at the 3’-end of tRNAox is responsible for the second crosslink with eRF1. To this end, we performed an experiment forming the binary complex “a”, and reduced the remaining aldehyde group with NaBH4 before eRF1 was bound to the A site. The result was the same as in lane 1 at pH 8.0 (Fig. 5C), showing that no crosslinked ternary complex was formed. It follows that after crosslinking of tRNAox with L36AL via one aldehyde group [7], the second one is involved in the crosslinking with eRF1.
The human translation termination factor eRF1 is not capable of binding and crosslinking with tRNAox in the absence of the ribosome
As shown in Figs. (5A and 5C) (lanes k), incubation of eRF1 with tRNAox in the absence of human 80S ribosome did not result in tRNAox crosslinking of the termination factor. Even at high concentration of tRNAox (20 µM) over that of eRF1 (2.5 µM), no tRNAox-eRF1 covalent complex was visible on the SDS-PAGE gel. Moreover, in a similar control experiment in the absence of ribosome but in the presence of the oligoribonucleotide GAA-GAC-UAA-AAA as mRNA [5 µM), [5’-32P]tRNAAsp76ox (10 µM) directed to the GAC codon was not capable of crosslinking with eRF1 (5 µM) directed to the adjacent UAA stop codon (results not shown). Furthermore, even when both the eRF1 factor and the 80S ribosome were present, the possibility that the “b” band formed (Figs. 5A and 5C) might represent a binary eRF1-tRNAox covalent complex with a calculated MW of 73,000 Da containing one molecule of eRF1 (48,000 Da) and one of tRNAox (25,000 Da) is ruled out by the following observations: (i) upon incubation of a given tRNA-binding protein with tRNAox in our experimental conditions for crosslinking, the obtained covalent protein-tRNAox complex, as followed by PAGE-SDS, always migrates in the gel with the expected higher molecular weight (in comparison with adequate protein markers) corresponding to the sum of the molecular weights of both macromolecules [9,16]. For example, on PAGE-SDS, the covalent complex between tRNAfMetox and E. coli methionyl-tRNAfMet transformylase migrated with an apparent molecular weight of 55,000 + 2,000 Da which agrees well with that predicted for one molecule of tRNA (25,000 Da) attached per molecule of enzyme (32,000 Da) [9]. Similarly, the covalent complex between tRNAfMetox (25,000 Da) and the trypsin-modified monomeric E. coli methionyl-tRNA synthetase (64,000 Da) migrated with an apparent molecular weight of 90,000 Da, consistent with the formation of a 1:1 covalent complex ([16] and unpublished results). Therefore, no change in the structure of proteins crosslinked to tRNAox has been ever observed that would be likely to affect their mobility on PAGE-SDS; (ii) in the presence of eRF1, formation of the “b” band is always correlated with the presence of the “a” band, while no “b” band was formed in the absence of eRF1, suggesting that the protein component that is common to the “a” and “b” bands is the L36AL protein.
The failure of eRF1 to crosslink with tRNAox in the absence of 80S ribosome contrasts with the case of the L36AL protein, which was shown to crosslink with tRNAox on and off the ribosome (Figs. 3 and 5C). These results suggest that, in contrast to RPL36AL, eRF1 binding to the A-site stop codon on the ribosome is a prerequisite for guiding and positioning of the GGQ region toward the CCA-arm of the P-tRNA.
At this stage, one should wonder whether the “b” band represents actually a ternary complex of P-tRNA with RPL36AL and eRF1. The presence of both L36AL and eRF1 in the 85,000 Da band “b'' (Fig. 5) was tested by MALDI mass spectrometry. First, we performed the crosslinking of tRNAox in the termination complexes sketched in Fig. (4). Second, the crosslinked products corresponding to the presumed ternary complex “b” (RPL36AL-tRNAox-eRF1) were separated by SDS-PAGE, followed by excision of the gel band “b” and in-gel digestion with trypsin. The mass spectra obtained by MALDI mass spectrometry of the tryptic digest were analyzed using a site-licensed Mascot database searching program. Protein scores greater than 29 were considered as significant, i.e. indicative of identity or extensive sequence homology when MS data are queried against a database consisting of human ribosomal proteins and eRF1. Upon analysis of the mass spectra and taking into account the detected large subunit ribosomal proteins having a molecular weight in the range 11,000-13,000 Da, it was possible to narrow the crosslinkable ribosomal protein to L30, L34, L36AL, and L37, among which L36AL and L37 are the best candidates owing to their scores (Table 2). The results in Table 2 show that eRF1, L36AL and L37 could be identified as belonging to the band under investigation, both ribosomal proteins being labeled in a mutually exclusive fashion.
MALDI mass spectrometric analysis of the tryptic digest obtained by in gel digestion with trypsin of the crosslinked product (gel band “b”) corresponding to the ternary complex RPL36AL-tRNAox-eRF1 (for details, see Materials and Methods).
MALDI mass spectrometric analysis of the ternary complex RPL36AL-tRNAox-eRF1.
| protein species | Molecular Weight (Da) | Score | pI |
|---|---|---|---|
| eRF1 | 49,228 | 36 | 5.51 |
| RPL30 | 12,947 | 4 | 9.65 |
| RPL34 | 13,513 | 3 | 11.48 |
| RPL36AL | 12,746 | 16 | 10.59 |
| RPL37 | 11,299 | 10 | 11.74 |
Since the score of a given protein in MALDI MS is the reflect of the efficiency of desorption of the related detected peptides, the low scores of the L36AL and L37 proteins, as compared with that of eRF1, might be explained by the difference of pI’s between eRF1 and these proteins (Table 2). In fact, owing to the basic pI (in the range 9.65-11.74) of the ribosomal proteins in Table 2, the peptides obtained by trypsin digestion would be basic as well, and would strongly interact with the acidic matrix used in MALDI mass spectrometry and their desorption would be problematic, and would lead to a dramatic underestimation of their scores. In sharp contrast to these aforementioned basic proteins, the score of 36 for the acidic protein eRF1 (pI of 5.51) would reflect a more efficient desorption of the acidic eRF1 peptides provoked by the repulsion between these peptides and the acidic matrix through their respective negative charges. Crosslinking of either L36AL or L37 with tRNAox in the “b” band could be explained by the assumption that, upon binding to the ribosomal A-site, eRF1 allows a second conformation of the ribosome and/or the tRNA, leading to a novel crosslink of the tRNA analogue to another large subunit ribosomal protein (namely L37) rather than to RPL36AL. These two large subunit ribosomal proteins have in common three properties. First, they belong to the corresponding L37e and the L44e protein families present in all eukaryotic and archaeal ribosomes [1, 2, 4]. Second, they belong to the family of large subunit ribosomal proteins having nonglobular extensions [1]. Third, they contain each a small globular domain with a zinc binding motif that coordinates a zinc ion [1]. In the crown view of the crystallographic structure of the large ribosomal subunit from Haloarcula marismortui (i.e. with the L1 stalk on the left), RPL44E and RPL37E, the archaeal orthologs of RPL36AL and RPL37, respectively, are located face to face at the subunit interface [1, 2]. Interestingly, the tips of their nonglobular extensions are located less than 10 Å from each other [1, 2], a distance that is compatible with the interaction of both extensions with the flexible periodate-oxidized CCA-end of tRNA. Intriguingly, while in the crown view of the large ribosomal subunit from yeast, RPL44E (or RPL42, the ortholog of RPL36AL) is located on the subunit interface [4], as in the archaeon [1, 2], RPL37 is located on the solvent-exposed side, in sharp contrast to its archaeal counterpart RPL37E. However, their respective locations [4] seem compatible with interactions of their extensions with the flexible CCA-end of the tRNA dialdehyde derivative.
Conformational Change of eRF1 Upon Binding to the Ribosome
It is interesting to note that a recently published cryo-electron microscopy study of the mammalian eukaryotic eRF1-eRF3-associated termination complex revealed that eRF1 must undergo substantial conformational changes to accomodate the GGQ motif into the PTC [17]. To test this possibility in the context of the crosslinking of eRF1 with tRNAox positioned at the P-site, we constructed 80S ribosomal complexes containing, in addition to a tRNAAsp76ox at the P-site and the eRF1 factor at the A-site, eRF3 in association with its substrate GTP or with GDPNP (the non-hydrolyzable GTP analog guanosine 5’-[β,γ-imido]-triphosphate; see Fig. 7). Analysis in 10% SDS-PAGE of crosslinked “a” and “b” bands corresponding to the binary complex RPL36AL-tRNAox and the ternary complex RPL36AL-tRNAox-eRF1, respectively, revealed that the “b” band is quenched in the presence of eRF3 independently of the nature of the guanine nucleotide (GTP or GDPNP) used (compare lanes 4 and 5 with lane 3 in Fig. 7). These results are consistent with a conformational change of eRF1 upon eRF3 binding on the ribosome and with the observation that eRF3 does not leave the ribosome after GTP hydrolysis [17]. Furthermore, quenching of the “b” band in the presence of eRF3 reflects the formation of eRF1-eRF3-GTP complex, in which eRF1 adopts a different conformation, as compared with the situation in the absence of eRF3. All in all, regarding the crosslinking of tRNAox with eRF1 on one hand, and with L36AL or L37 on the other hand, one can imagine that the P/E tRNAox forms a crosslink with L36AL or L37, then rearranges before crosslinking with eRF1 which would be also subjected to some conformational change, as discussed above. Finally, for unknown reasons, the intensities of the “a” bands (RPL36AL-tRNAox complex) were also affected by the binding of eRF3 to eRF1 (compare lanes 4 and 5 with lane 3 in Fig. 7). Another observation from Fig. (7) is that the yields of crosslinking of “b” band containing the ternary complex RPL36AL-tRNAox-eRF1 is similar in the absence or presence of an E-site tRNA (compare lanes 2 and 3 in Fig. 7). As native tRNAAsp was recently shown to bind easily to the E-site of the human large ribosomal subunit [7], these results might be interpreted as follows: In the presence of an E-tRNA the neighboured tRNAox cannot adopt the hybrid state P/E but rather the classical P/P site. A crosslink of L36AL with tRNAox in this situation shows that this ribosomal protein lies closer to the P site than previously reported [7]. Finally, the human 80S ribosome in a complex with P-site bound tRNAox and with A-site bound eRF1 used here mimics the post-termination state of the ribosome. Thus the results shed light onto the mutual positioning of the P-tRNA, L36AL and eRF1 at the end of the translation process.

Analysis in 10% SDS-PAGE of crosslinked ribosomal complexes 1-5 obtained in the presence of [5’-32P]tRNAAsp76ox (lanes 1-5, respectively). Autoradiogram of the gel. The positions of tRNA analogues and crosslinking products are marked.
pH Dependence of the Crosslinking of Endogenous Human Ribosomal L36AL Protein with [5’-32P]tRNAAsp75ox
The strong increase of the crosslinking yields at pH 8.0 (Fig. 5B) prompted us to follow the crosslinking reaction between RPL36AL and [5’-32P]tRNAAsp75ox as a function of the pH of the incubation mixture. This truncated tRNAox species was previously found to crosslink to Lys-53 of RPL36AL through the formation of a reversible Schiff base between the ε-amino group of Lys-53 and the 2’, 3’ aldehyde groups of tRNAox [7]. Fig. (8) shows the formation of the reversible Schiff base and its irreversible reduction with sodium borohydride (NaBH4) or with sodium cyanoborohydride (NaBH3CN) [16]. The equilibrium of the Schiff base is frozen by reduction with sodium borohydride (NaBH4) which simultaneously reduces the Schiff base and the unreacted tRNA dialdehyde, while NaBH3CN is capable of continuously displacing this equilibrium by specifically reducing the imminium group and leaving intact the reacting 2’, 3’ aldehyde groups of tRNAox [16]. In the experiment of Fig. (5B), the strong reducing agent NaBH4 was preferred to the latter mild one, in order to trap the fraction of unprotonated reactive ε-amino group that is only responsible for the rate-limiting nucleophilic addition to the aldehyde to form the Schiff base (Fig. 8). Therefore, the crosslinking yield at a given pH in the presence of NaBH4 reflects the fraction of unprotonated reactive ε-amino groups.
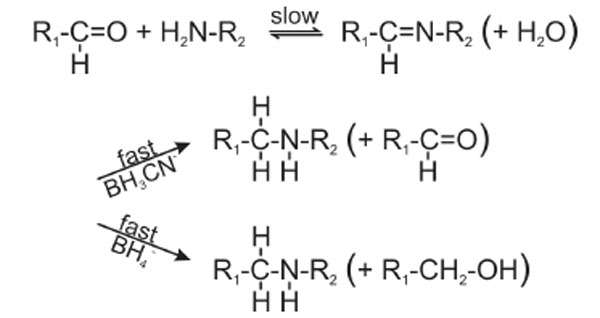
Reaction scheme of the Schiff base formation and its in situ reduction with sodium borohydride (NaBH4) or sodium cyanoborohydride (NaBH3CN). With both reducing agents, the overall crosslinking reaction is rate-limited by the formation of the imminium moiety, not by its in situ reduction.
Assuming that the maximal yield of crosslinking in the binary RPL36AL-tRNAox complex “a” was obtained at pH 8.0 and thus considered as 100%, the yields of crosslinking for the “a” band from Fig. (5B) (lanes 1 to 6) were 10% at pH 5.0, 23% at pH 6.0, 50% at pH 7.0, 67% at pH 7.5, 100% at pH 8.0 and 90% at pH 9.0. These data were then used to plot the crosslinking yield versus increasing pH values. As shown in Fig. (9), the crosslinking yield is proportional to the pH, i.e. to the mole fraction of a basic group with a pKapp of about 6.9 describing the dissociation of the protonated form of the reactive ε-amino group of Lys-53. This value of pKapp is 3 to 3.5 units lower than is normally found for an ε-amino group of a lysyl residue in a protein, and it is likely that this abnormally low pKapp accounts for the reactivity of Lys-53 through the Schiff base formation leading to crosslinking. In conclusion, the abnormally low pKapp of the ε-amino group of Lys-53 would result in a greater mole fraction in the reactive, unprotonated form at neutral pH values, and consequently a greater crosslinking yield. Since, in general, unusually reactive side chains are also those required for biological activity, so that pH dependence of activity can be correlated with pH dependence of the modification reaction, it is most probable that the ε-amino group of Lys-53 with a pKapp of 6.9 might be of functional importance for the biological activity of human 80S ribosome. Even though at pH near 5.0 the human 80S ribosomes can be inactivated due to precipitation, the mole fraction in the reactive, unprotonated form of the crosslinked residues and consequently the crosslinking yields would not be affected.
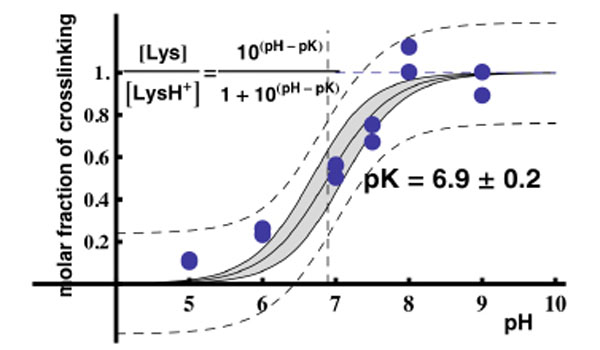
Plot of the molar fraction of crosslinking as a function of pH. Two data sets from two separate experiments of Fig. (5B) were used to obtain this graph. The data were fitted with Mathematica to the function inscribed in the figure with the following features : the dashed lines defines the 95% confidence intervals for the prediction of a single value, whereas the continous lines and the gray-shaded region define the 95% confidence intervals for the prediction of the average curve with pK = 6.9 ± 0.2.
Checking the peptidyl-tRNA Hydrolase Activity of the Recombinant Human L36AL Protein
The presence of the unusually reactive Lys-53 in proximity to the universally conserved methylated GGQ motif common to translation termination factors (RFs) tempted us to speculate that the GGQ motif of RPL36AL is implicated in promoting peptidyl-tRNA hydrolysis as does the same motif in the RFs [19-21] and in proteins that display peptidyl-tRNA hydrolysis activity [22-24]. We addressed this question by measuring the peptidyl-tRNA hydrolysis activity of isolated recombinant RPL36AL, in comparison with that of the enzyme peptidyl-tRNA hydrolase (Pth) from E. coli, by using N-acetyl[3H]Phe-tRNAPhe as a substrate. As shown in Fig. (10), no Pth-like peptidyl-tRNA hydrolase activity was detectable under experimental conditions used for the Pth enzyme.
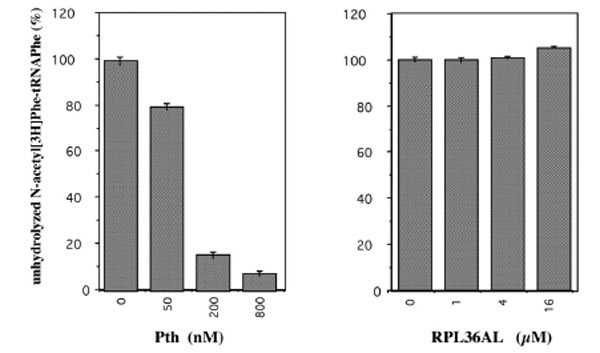
Peptidyl-tRNA hydrolase assay of the recombinant human L36AL protein using N-acetyl[3H]Phe-tRNAPhe as a substrate. The percent of residual N-acetyl[3H]Phe-tRNAPhe precipitable in trichloracetic acid was measured as a function of the concentration of the recombinant human RPL36AL. As a control, the activity of E. coli peptidyl-tRNA hydrolase (Pth) as a function of enzyme concentration is also shown.
DISCUSSION
Models of the 3-D Structure of the Human L36AL Protein Alone or in Complex with tRNA
Models of the 3-D structure of the human L36AL protein alone or in complex with tRNA (Fig. 1) predict specific interactions reminiscent of the general model of tRNA-aminoacyl-tRNA synthetase interaction, which shows that synthetases bind their cognate tRNAs along and around the inside of the L-shaped structure, recognizing the acceptor stem including the CCA arm and often also the anticodon [14,15]. Binding assays on Biacore using isolated recombinant purified human RPL36AL protein and tRNA or truncated tRNAs reveal that these interactions are characterized by strong binding affinities in the nanomolar range, which argues for a specific interaction. However, it is well known that ribosomal proteins generally bind to structured regions of rRNAs with high affinity. RPL36AL is the only ribosomal protein that can intereact with tRNA on and off the ribosome.
Interestingly, recent structural data on bacterial 70S ribosomes from Thermus thermophilus have revealed that, at least in bacteria, proteins play a supporting role in peptide-bond formation and are closer to the active site than previously reported [25]. These data have provided direct structural evidence for the interactions between the proteins L16 and L27 and the A- and P-site tRNAs [25]. In addition, previous biochemical studies had demonstrated that, in solution, E. coli L16 protein interacts with tRNA with a KD value of 5 X 10-7 M [26, 27]. We note that specific interactions between aminoacyl-tRNA synthetases and their cognate tRNA had been shown to be accounted for by KD and/or KM values in the micromolar range [28], which are about 2-3 orders of magnitude higher than that (in the nanomolar range) of tRNA binding to L36AL protein reported in Table 1, suggesting that the latter complex is formed with stronger binding affinities than those involving aminoacyl-tRNA synthetases (or protein L16) and tRNA. Likewise, Biacore had been used to obtain detailed insight into interactions between RNA and non-ribosomal proteins carrying RNA recognition motifs, and KD values in the nanomolar to picomolar range were deduced [29] as in the present report.
The fact that the binding affinities for L36AL protein of tRNA species having variable lengths at the 3’-end are of the same order of magnitude suggests that the contribution to the interaction with RPL36AL of the CCA region of tRNA is rather weak, and that other regions of the tRNA molecule are actually involved in the formation of stable RPL36AL:tRNA complexes. One possibility is that the consensus pattern 61Kx(TorV)KKxxL(KorR)xxC72 of the r-proteins of the L44e family might be involved in the interaction with tRNA around the inside of the L-shaped structure (Fig. 1B). It should be noted that the presence of a conserved sequence in all the eukaryal and archaebacterial L44e-family members suggests that this protein region is of functional significance and might participate in the interaction of the protein with tRNA or 28S (23S) rRNA or both.
As a result of multiple sequence alignment of the r-proteins of the L44e family, we found a nucleotide binding motif corresponding to fragment 86KRCKMeHFELGGD96 of RPL36AL that is strictly conserved in all eukaryotic organisms, and that had been observed for the first time in human ribosomal protein L7 [30], the ortholog of the bacterial L30 protein (Fig. 1C). This motif named nucleotide binding domain 2 (NBD2) by von Mikecz et al. [30] is conserved in all eukaryotic RPL7, and was shown to bind preferentially to 28S rRNA [30]. As shown in Fig. (1C), the consensus pattern between eukaryotic RPL7 and L36A/L44 proteins is KK(orR)xxHF(orY)xxGGD(orE). The presence of the rare amino acid His, the nucleotide binders Lys, Arg, Phe and Tyr residues and the loop-forming Gly-Gly dipeptide, makes it likely that this motif might represent a new class of RNA binding domain as proposed by von Mikecz et al. [30] (Fig. 1C). Finally, all known eukaryotic RPL36A and archaebacterial RPL44E contain zinc binding motifs that share the potential to coordinate a zinc ion. This metal ion (Fig. 1A) is supposed to stabilize the structures of these RNA-binding proteins and/or to be coordinated at the protein-RNA interface, as part of the recognition mechanism. Collectively, the data in the present report suggest interactions of the L36AL protein with both tRNA and rRNA.
Crosslinking Studies on the Human Recombinant Protein L36AL
Crosslinking of the P-site tRNAox with proteins occurs through the formation of a reversible Schiff base between the 2’,3’-aldehyde groups of tRNAox and the (ε-amino group of a lysyl residue [6], the covalent linkage being stabilized by reduction of the Schiff base with sodium cyanoborohydride (NaBH3CN) [31,32]. Therefore, crosslinking of recombinant RPL36AL with tRNAox met all the criteria of an affinity labeling reaction, similarly to all tRNAox crosslinking reactions performed so far [6, 9, 10, 16, 31, 32]. The crosslinking reaction resulted in a 1:1 covalent protein-tRNAox complex. In all tRNAox crosslinking experiments performed so far, the stoichiometry tRNA:protein (enzyme or a ribosomal protein) was always found equal to the stoichiometry known for the active complex [9, 16, 31, 32] indicating the physiological relevance of the observed complexes.
The crosslinking yield with tRNA76ox in the absence of eRF1 is weaker than those with the shortened tRNAox species (Fig. 3), in agreement with previously reported crosslinking data involving the endogenous ribosomal protein L36AL in situ [7]. The amino acid residues of the recombinant protein crosslinked with tRNAox in a binary complex were not identified in the present report. However, there are only few residues that are reactive toward the aldehyde groups of the tRNA dialdehyde derivative [7, 10, 31, 32]. Candidates are lysyl or arginyl residues that exhibit abnormally low pKapp values, resulting in a large mole fraction in the reactive, unprotonated form at neutral pH values, and consequently in a large crosslinking yield. This is valid for Lys-53, and therefore, Lys-53 is most probably the crosslinked residue, as we previously reported in the case of RPL36AL within the ribosome [7]. Our data represent the first biochemical evidence for specific interactions between a tRNA and a eukaryotic ribosomal protein (namely RPL36AL) on and off the ribosome.
The CCA-arm of P-site Bound tRNA is in Proximity with Both the Ribosomal L36AL Protein and the Termination Factor eRF1
When the termination factor eRF1 is bound to the A-site in the presence of the stop codon UAA, a P-site bound tRNAox is capable of crosslinking with both the ribosomal protein L36AL and eRF1, giving rise to the formation of a ternary covalent complex RPL36AL-tRNAox-eRF1. The molecular weight of the ternary complex corresponds to a 1:1:1 stoichiometry (Figs. 5 and 6). eRF1, L36AL and L37 could be identified as belonging to the “b” band representing a ternary complex, both ribosomal proteins being labeled in a mutually exclusive fashion. Crosslinking of either L36AL or L37 with tRNAox could be explained by the assumption that, upon binding of eRF1 to the ribosomal A-site, the ribosome and/or the tRNA adopts two conformations, one of which leads to a novel crosslink of the tRNA analogue to the ribosomal protein L37. It is conceivable that flexible regions of all partners are involved in the crosslinking, i.e. the comparably flexible GGQ domains of eRF1 and of RPL36AL, as well as the CCA-arm of P-tRNAox.
A Possible Model for the Interactions Between RPL36AL, the CCA-arm of P-tRNA and eRF1, at the Active Site of Human 80s Ribosome
Now we are in the situation to propose a model for the interactions between the ribosomal L36AL protein, the CCA-arm of P-site bound tRNA and the translation termination factor eRF1 bound to an A-site stop codon (Fig. 11), taking into account recently published structural and photocrosslinking data as well as the chemical crosslinking results reported here. We have recently proposed that the tRNAox species used in the present report might bind first to the classical P/P site before they flip spontaneously into the P/E site, where they are sampled by the crosslinking with Lys-53 of RPL36AL [7], in agreement with recent cryo-electron microscopy and single-molecule FRET data [33]. Our crosslinking data agree well with photocrosslinking data using a tRNAAsp analogue substituted with 4-thiouridine in position 76 (tRNAAspp-s4U76), which could be cross-linked to C4335 located very close to Lys-53 of RPL36AL in the E-site region of human 80S ribosome [18]. Crosslinking of the CCA-arm of a P-tRNA with both RPL36AL and eRF1 implies that the P-tRNA is present in the P/P state, while the L36AL protein due to the flexibility of its loop extension is capable of reaching the P-site, where it could interact with the CCA-arm and the acceptor stem of tRNA. Accordingly, we propose the model in Fig. (11), where the CCA end of the P-tRNA is sandwiched between the methylated GGQ motifs of RPL36AL and the A-site bound eRF1. In this model, the GGQ loop of eRF1 faces nucleotide A76 of P-tRNA, since at the end of the translation process, this nucleotide which bears the ester carbone of the peptidyl-tRNA is the natural target of the GGQ motif of eRF1 for the nucleophilic attack (via a water molecule) leading to the release of the polypeptide chain. Therefore, this model is compatible with the observation that the crosslinking yields of the ternary complexes “b” decrease as a consequence of the removal of A76 of the CCA-arm of tRNA. Decreasing of the crosslinking yield of the ternary complex “b” upon removal of A76 is in striking contrast to the crosslinking yields of the binary RPL36AL-tRNAox complexes “a”, which shows higher yields with tRNAox lacking the CCA-end. The latter results would suggest that the interaction with RPL36AL of the CCA region of tRNA is rather weak, and that other regions of the tRNA molecule are actually involved in the formation of a binary RPL36AL:tRNA complexes, as discussed above. One possible explanation is that, in the absence of eRF1, the CCA end of tRNA might be dispensable for the interaction between RPL36AL and the tRNA, while in its presence, the ribosomal protein makes specific contacts with the CCA end. This agrees well with the fact that the tRNA CCA-end must be in full-length for crosslinking both RPL36AL and eRF1.

Model for the interactions between the ribosomal protein L36AL, the CCA-arm of P-site bound tRNA and the translation termination factor eRF1 bound to an A-site stop codon.
The possibility that the GGQ motif of RPL36AL is implicated in promoting peptidyl-tRNA hydrolysis as the same motif in eRF1 [19-21] and in proteins that display peptidyl-tRNA hydrolysis activity [22-24] could not be confirmed with isolated recombinant RPL36AL that did not show any hydrolase activity under condtions used for the enzyme peptidyl-tRNA hydrolase (Pth) from E. coli. However, since most of the GGQ-containing proteins hydrolyze peptidyl-tRNA on the ribosome, it is possible that RPL36AL catalyzes peptidyl-tRNA hydrolysis on the ribosome in some unusual conditions and fashion.
Structure comparison around the GGQ motif (Fig. 12) of eRF1 from various species suggests that Lys-178 or Lys-179 are probable sites crosslinkable with tRNAox. Nearby Arg residues are less likely, because it is well known that arginine residues are by far less reactive than lysine residues, as far as Schiff base formation with aldehyde groups is concerned. Work is in progress to identify the residues involved.
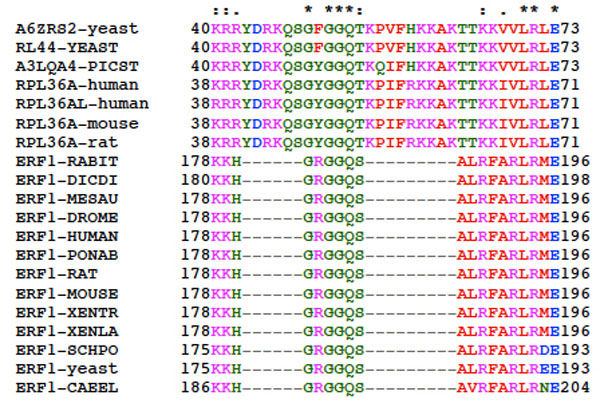
Multiple sequence alignment of eRF1s in the region encompassing the GGQ motif common to all class-1 translation termination factors and the corresponding region of rPs of the RPL36A/RPL44E family from different eukaryotic organisms. The top line labels residues as either strictly conserved (*), highly conserved (:) or weakly conserved (.). The alignment was generated with the program ClustalX. XENTR, Xenopus tropicalis; CAEEL, Caenorhabditis elegans.
MATERIALS AND METHODS
Modeling of the Human RPL36AL three-dimensional Structure and of Its Interaction with tRNA
Comparative modeling of the RPL36AL structure was achieved using as template the structure of the Haloarcula marismortui large ribosomal subunit chain 3 at 2.2 Å (PDB: P32411). This template was identified by hhsearch [34], the resulting alignment has 21 % sequence identity. 3-D model generation was performed using modeler [35] to generate 50 models with only slight variations. Not surprisingly, the models are close to the corresponding structure obtained in a previous study by Chandramouli and co-workers [13], who modeled the structure of the mammalian 80S ribosome from cry-electron microscopy data. Subsequent structural alignments were performed using fr-tmalign [36]. The overlaid structures of RPL36AL and the tRNA correspond to that of the ribosomal protein modeled in the present report and that of free tRNAPhe (PDB ID 1JGQ).
Biological Materials
40S and 60S ribosomal subunits with intact rRNAs were isolated from unfrozen human placenta [37]. Prior to use, the subunits were re-activated by incubation in binding buffer A (50 mM Tris-HCl, pH 7.5, 100 mM KCl, 10 mM MgCl2 and 0.5 mM EDTA) at 37°C for 10 min. 80S ribosomes were obtained by association of the re-activated 40S and 60S subunits used in an equimolar ratio. Activity of the ribosomes in the poly(U)-directed binding of [14C]Phe-tRNAPhe was 70%. E. coli 70S ribosomes were performed as in reference [38]. The mRNA analogues poly(U) and GAA-GAC-UAA-AAA, as well as E. coli tRNAPhe were purchased from Sigma-Aldrich. tRNAAsp from yeast was purified by conter-current chromatography, followed by separation by polyacrylamide gel electrophoresis. L-[14C(U)]Phenylalanine (18GBqxmmol-1) was from Perkin Elmer. [14C(U)]Phe-tRNAPhe was obtained as in reference [38]. Purified elongation factors from calf brain EF-1a, EF-1b, EF-2 were obtained as in reference [39]. E. coli elongation factors EF-Tu, EF-Ts and EF-G were purified as described [40]. Poly(Phe) synthesis in the eukaryotic system was determined as incorporation of L-[14C(U)] Phenylalanine into hot trichloroacetic acid-insoluble material, as described in reference [39]. The poly(Phe) synthesis assay in the prokaryotic system contained 0.4 microM EF-Tu EF-Tu, 0.2 µM EF-Ts, 0.2 microM EF-G and 0.3 µM E. coli 70S ribosomes. tRNAAsp species shortened at the 3’-end and lacking either the A76 (tRNAAsp75), the C75-A76 (tRNAAsp74), the C74-C75-A76 (tRNAAsp73), the G73-C74-C75-A76 (tRNAAsp72) or the A72-G73-C74-C75-A76 (tRNAAsp71) nucleotides were obtained stepwise by combining periodate, lysine, and alkaline phosphatase treatments as previously described [7]. Homogeneity of these truncated tRNAs was probed by high-resolution gel electrophoresis on 10% polyacrylamide gel containing 8 M Urea and Tris/borate/EDTA, where each of the modified species appeared not damaged and homogeneous (results not shown). Gel was stained with “Stains all” (Sigma). 32P-labeling of tRNA analogues at the 5’-end was carried out after dephosphorylation by alkaline phosphatase (Roche), in the presence of γ-[32P]ATP and polynucleotide kinase. Periodate-oxidized tRNAs (tRNAox) were prepared as described previously [16].
Expression and Purification of the Recombinant L36AL Protein from Homo Sapiens
His-Tagged RPL36AL was expressed in Solu BL21 E. coli cells (AMS Biotechnology) harboring the plasmid pColdI-RPL36AL. A fresh overnight culture was used to inoculate 5 liters of LB with 50 µg/ml carbenicillin and grown with shaking at 37°C to 0.5 OD600, after which induction with 0.4 mM isopropyl-ß-D-thiogalacto-pyranoside took place at 15°C with incubation up to 30 hours. After harvest, the cells (8.5g) were washed in PBS, sonicated 15 times for 10 s at 4°C in 40 ml buffer A (25 mM Tris-HCl pH7.5, 0.5 M NaCl, 10% glycerol, 10 mM Imidazole, 1 mM ß-ME) containing 0.05% Tween 20, 1 mM MgCl2, 0.5 mg/ml lysosyme, 100 µg/ml DNase, one tablet of complete mini EDTA free protease inhibitor cocktail (Roche Diagnostics), and centrifuged (100,000 x g for 30 minutes). Western blot analysis on supernatant and pellet using penta.His biotin conjugate (Qiagen) revealed that RPL36AL was nearly insoluble. Consequently, the pellet was suspended in 30 ml buffer A containing 8 M urea and sonicated 30 times for 6 s. The supernatant obtained by centrifugation (100,000 x g for 30 minutes) was applied two times to Ni-NTA superflow (Qiagen) column (2 ml), and washed with 20 ml buffer A containing 8 M urea and 25 mM Imidazole. The protein was then eluted with 10 ml buffer A containing 8 M urea and 0.4 M imidazole. This fraction was concentrated to 1.5 ml in an Amicon ultrafiltration apparatus (Millipore membrane PM10) and applied to Hi-load 16/60 Superdex 75 column (GE healthcare) equilibrated in buffer B (50 mM Tris-HCl pH7.5, 0.35 M NaCl, 7 mM ß-ME). Fractions eluted containing purified RPL36AL (5 mg), free of urea were concentrated by ultrafiltration and dialyzed against buffer B with 50% glycerol and stored at -25°C.
Cloning, Expression and Purification of the Human eRF1
The full length cDNA encoding human eRF1 with the C-terminal His6-tag fusion was cloned into pET23b(+) vector (Novagen) as described [41]. eRF1 was expressed in E. coli, strain BL21(DE3) and purified by combination of affinity chromatography on Ni-NTA resin, Superflow (Sigma) as described [41] and ion-exchange chromatography. eRF1 was loaded onto 1 ml HiTrapQ column (GE Healthcare) equilibrated with buffer A (100 mM KCl, 20mM Tris-HCl, pH 7.5, 6 mM β mercaptoethanol, 5% glycerol), the column was washed with buffer A (10 ml) and the linear gradient of KCl (0.1M → 1M) in buffer A is used for elution of eRF1. The volume of gradient was equal to 20 ml, the fraction volumes were 0.5 ml (flow rate 1ml/min). Fractions were analyzed by 10% SDS-PAGE, the eRF1 peak fractions were combined, dialyzed against buffer A and purified eRF1 was stored at -70°C.
Biosensor assay
The surface plasmon resonance (SPR) biosensor experiments were performed on a Biacore 3000 instrument (GE Healthcare). The purified His-tagged L36AL protein was immobilized on NTA sensor chip (GE Healthcare) to reach an immobilization level of the protein of 5,000-7,000 resonance units (RU), according to the manufacturer’s recommendations. The protein and the tRNA species were diluted to a final concentration of 1 µM in HBS-EP GE Healthcare (10 mM HEPES pH 7.4, 150 mM NaCl, 3 mM EDTA, 0.005% surfactant P20) containing 2 mM MgCl2. A reference surface without protein was prepared using the same procedure. Evaluation of non-specific background signals was performed by using tRNAAsp on NTA chips uncoated with His-tagged RPL36AL. Running buffer (10 mM HEPES pH 7.4, 150 mM NaCl, 3 mM EDTA, 0.005% surfactant P20) and binding buffer (10 mM HEPES pH 7.4, 150 mM NaCl, 2 mM MgCl2) were used. Between two binding assays at two different concentrations, the surface was regenerated by injection of 50 mM NaOH at a flow rate of 10 µl/min and 30 s contact time, followed by a 10 min dissociation by washing at a continuous flow of the running buffer. The experimental data from individual kinetic binding experiments were overlaid and analyzed using BIAevaluation 4.1.F46 Bindi software (GE Healthcare) with 1:1 binding model.
Crosslinking of tRNA Analogues to 80S Ribosomes or to the Purified Recombinant L36AL Protein
Crosslinking of tRNA analogues to the purified recombinant L36AL protein was performed as follows: 200 pmol of RPL36AL were incubated for 30 min at 37°C with 200 pmol of the tRNAox species in 20 µl 50 mM Tris-HCl buffer (pH 8.0) containing 10 mM MgCl2 and 5 mM sodium cyanoborohydride (NaBH3CN). The mixtures were applied onto a 10% polyacrylamide gel run by 8 M urea electrophoresis and colored with Stains all. Crosslinking of the tRNAox species to ribosomes was performed as described earlier [6]. Reaction mixtures (20 µl each) contained 10 pmol of 80S ribosomes, 100 pmol of oligoribonucleotide GAA-GAC-UAA-AAA as mRNA and 20 pmol of each tRNAox species in buffer A (50 mM Tris-HCl at the indicated pH, 100 mM KCl, 10 mM MgCl2 and 0.5 mM EDTA) with incubation for 40 min at 25°C, for the formation of complexes 1. For the crosslinking studies as a function of the pH of the incubation mixture, we used binding buffers A5.0, A6.0, A7.0, A7.5, A8.0 or A9.0 which contained 50 mM Tris-HCl at a pH of 5.0, 6.0, 7.0, 7.5, 8.0 or 9.0, respectively. In these cases, sodium borohydride (NaBH4; 50 mM final concentration) was used as a strong reducing agent instead of NaBH3CN, in order to trap the fraction of unprotonated reactive ε-amino group that is only responsible for the nucleophilic addition to the aldehyde to form the Schiff base. Complexes 2 were obtained by addition to complexes 1 of 50 pmol of human eRF1 at the indicated pH, followed by incubation in the same conditions as above. In a control experiment in the absence of ribosome but in the presence of the oligoribonucleotide GAA-GAC-UAA-AAA as mRNA (5 µM), [5’-32P]tRNAAsp76ox (10 µM) directed to the GAC codon was not capable of crosslinking with eRF1 (5 µM) directed to the adjacent UAA stop codon. In other controls in the presence of eRF3 associated with its substrate GTP or with GDPNP (guanosine 5’-[β,γ-imido]-triphosphate), 50 pmol of the protein and of the nucleotide were added to the incubation mixtures. Crosslinked proteins were analyzed by SDS-PAGE on 10% polyacrylamide gels, and crosslinking yields were calculated as described earlier [7].
Purification of Pth and Deacylation Assay
Pth was expressed in E. coli BL21(DE3) transformed with pET15blpa-Pth (a gift from Dr. Pierre Plateau). A fresh overnight culture was used to inoculate 0.8 liter of LB with 50 µg/ml ampicillin and grown with shaking at 37°C to 0.8 OD600, after which induction with 0.1 mM isopropyl-ß-D-thiogalacto-pyranoside took place at 18°C with incubation up to 20 hours. After harvest, the cells (6 g) were washed in PBS, sonicated 15 times for 10S at 4°C in 30 ml buffer A (25 mM Tris-HCl pH7.5, 0.5 M NaCl, 10% glycerol, 10 mM Imidazole, 1 mM ß-ME) containing 0.05%Tween 20, 1 mM MgCl2, 0.5 mg ml-1 lysosyme, 100 µg ml-1 DNase, one tablet of complete mini EDTA free protease inhibitor cocktail (Roche Diagnostics), and centrifuged (100, 000 x g for 30 min). The supernatant. was applied to Ni-NTA superflow (Qiagen) column (2 ml), and washed with 20 ml buffer A containing 50 mM Imidazole. The protein was then eluted with 10 ml buffer A containing 0.4 M imidazole. This fraction was concentrated in an Amicon ultrafiltration apparatus (Millipore membrane PM10) and dialyzed against 50 mM Tris-HCl pH 7.5, 1 mM MgCl2 0.1 M NaCl, 50% glycerol, 7 mM ß-ME and stored at -25°C. tRNAPhe from Sigma was aminoacylated using [14C]phenylalanine ( 1.2 Ci/mmol) using phenylalanine-tRNA synthetase [42] and acetylated by the method of Haenni and Chapeville [43]. Deacylation assay was determined in 15 µl containing 25 mM Tris-HCl pH7.5, 10 mM MgCl2 ,0.1mM EDTA, 1 µM N-acetyl [14C]PhetRNAPhe and the indicated concentrations of Pth or RPL36AL. Reaction was initiated with addition of the enzyme. After 12 min incubation at 30°C, 12 µl samples were taken and spotted onto glass fibre filters Whatman GFC and cold 10% trichloroacetic-insoluble material on the filters was measured.
MALDI Mass Spectrometric Analyses
The MALDI mass spectrometric analyses were performed as described by Sharifulin et al. [44]. The ternary complex RPL36AL-tRNAox-eRF1 was prepared as under Materials and Methods. Bands “b” corresponding to the 85 kDa crosslink were excised from Coomassie-stained gels. The gels were cut into small pieces and destained by washing with NH4HCO3 (25 mM) containing CH3CN (50%). Proteins were reduced in-gel by incubation (56°C, 1 h) with DTT (10 mM) in 25 mM NH4HCO3, followed by alkylation of cysteine residues with 55 mM iodoacetamide in 25 mM NH4HCO3 during 45 min in the dark. Proteins were digested by incubation (37°C, overnight) with trypsin (30 µg.mL-1; Promega, sequencing grade). The peptides were desalted on a ZipTip C18 column (Millipore) then eluted with TFA (4 µL; 0.1% in 50% CH3CN) and subjected to MALDI TOF MS. Samples (1 µL) were mixed with MALDI matrix material (1 µL; saturated solution of 3,5-dimethoxy-4-hydroxycinnamic acid in acetonitrile/TFA (0.1%) 1:2, v/v). Mass spectra were obtained on an Autoflex speed series MALDI-TOF mass spectrometer (Bruker Daltonics) equipped with a pulsed N2 laser (337 nm) in a positive reflectron mode. Ions formed by the laser beam were accelerated to 23 keV. The final spectra were obtained by accumulating of ~1500 single laser shots. External calibration in positive mode was done by using Peptide Calibration Standard II (#222570, Bruker Daltonics). Mass accuracy of ~15 ppm was usually achieved. Mass spectra were processed with flexAnalysis 2.4 software (Bruker Daltonics). Peptide mass calculations were performed by using the protein utilities at http://www.expasy.org. The spectra (peaks) were analyzed by using a site-licensed Mascot database searching program (200 ppm tolerance; Matrix Science, London, UK). Protein scores superior to 29 were considered significant, that is, these indicate identity or extensive sequence homology (p<0.05) when MS data were queried against a database consisting of human ribosomal proteins and eRF1.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
We are grateful to Professors Jean Cognet and Yannick Andéol for insightful advises, and to Drs. Arsène P. Der Garabedian, Joël Pothier and Gérard Keith for fruitful discussions. We gratefully thank Mathilde Sayegh and Jilali Dalouz for technical assistance, Jean-Pierre Le Caër for the mass spectrometric facilities, and Drs. Cheikhe Hadjibou Soumare, Guy-Amédée Ajanohoun, Mahamane Hamidine and Safiatou Ouattara for their constant interest for this work.

