All published articles of this journal are available on ScienceDirect.

Effect of Target Length on Specificity and Sensitivity of Oligonucleotide Microarrays: A Comparison between Dendrimer and Modified PCR based Labelling Methods
Abstract
DNA microarrays are widely used as end point detectors for gene expression analysis. Several methods have been developed for target labelling to enable quantification but without taking target length into consideration. Here we highlight the importance of choosing the optimum target length that would ensure specificity without compromising sensitivity of the assay. For this, eight plasmids that are identical to each other except for a closely related 23 bp unique reporter (UR) sequence were used to examine the hybridization efficiency for these URs. Targets of various lengths were generated and labelled as follows: full length and 330 bases transcripts using a dendrimer labelling method, 120 bp amplicons by the modified PCR end labelling method and synthetic labelled targets of 33 bases. This report also shows the advantages of using the modified PCR method over other labelling methods in generating labelled amplicons of the desired lengths to maximize hybridization efficiency.
1. INTRODUCTION
DNA microarrays are a powerful technology that has versatile applications in many fields of life science research. One of the major areas that this technology has been applied to is the determination of relative and absolute RNA abundance for genetic expression analysis purposes [1-3]. DNA microarrays can explore transcriptional and genomic profile for thousand of genes in a single experiment. The ability to investigate genome wide functions quickly and efficiently made it a widely used and successful tool. It has also been used successfully for DNA sequencing [4,5], SNP genotyping [6,7], gene discovery [8,9], disease diagnostic [10-11] and pathway reconstructions [12,13].
Oligonucleotide and cDNA microarrays are the two major categories of DNA microarray [14]. The former is composed of single stranded 25-70 mers which are either pre synthesized and immobilized or synthesized in situ. The latter uses double stranded DNA products that usually range from 0.6 to 1.5 kbp. They are generated from PCR amplification of cDNA library clones or genomic DNA [15]. cDNA microarrays are the method of choice when the genome sequence is not available [16]. Besides the quantity and quality of captures and targets, several parameters such as substrate quality, spotting buffer, labelling method, hybridization condition and scanning procedure affect the quality of datagenerated from the probing of DNA microarrays with biological samples.
In a single array, thousands of cDNAs or oligonucleotides are spotted at high densities on a solid surface. Glass is the main surface of choice. It is cheap, chemically inert, transparent, and resistant to high temperature, with low intrinsic fluorescence, and can be functionalized efficiently with many functionalities including epoxy [17], amine [18], aldehyde [19] or poly L-Lysine [20] groups. The positively charged amino group of amine and poly L-Lysine coated slides adsorb negatively charged phosphate groups of DNA through electrostatic interactions. This non covalent binding nature makes DNA films susceptible to removal under high temperature or high salt conditions [21]. Slides therefore are exposed to UV to ensure cross linking with DNA thymidine residues [16,22,23]. It has been reported that these slides, especially poly L-Lysine, suffer from aging problems [24,25]. Aldehyde coated slides bind specifically to DNA through Schiff base formation which is then reduced with NaBH4 to form a stable secondary amine. However, they are limited to capture molecules that contain or are modified with an amine group. On the other hand, the ability of epoxy coated slides to immobilize either modified or unmodified capture molecules make them versatile and cost effective, and they are widely used. Importantly, this reactive surface forms a stable covalent bond through a nucleophilic attack of DNA primary amine groups [16,26].
Oligonucleotide capture arrays need to be designed carefully to optimize the sensitivity and specificity of the assay. Sensitivity refers to the maximum binding of an oligonucleotide to a corresponding target sequence with a perfectly matching sequence (PM), while specificity represents concomitant minimal binding to non target sequences with mismatches (MM) [27]. It became common in microarrays to include MM derivatives for each PM oligonucleotide to serve as controls. MM captures are usually identical to the PM with the exception of one nucleotide mismatch towards the middle of the sequence [28,29].
Several parameters such as temperature, formamide and monovalent salt concentration, time and washing protocol, and other factors such as the formation of secondary structure, affect the hybridization process [16,30]. Increasing temperature decreases non specific binding, but excessive temperature will decrease sensitivity. Therefore the hybridization temperature is carefully chosen to allow maximum specificity without compromising sensitivity [16]. Generally, maximum hybridization is achieved 20-25 °C below the melting temperature (Tm) [31]. Each increase of 1% in formamide concentration has been found to decreases the Tm by an average of 0.63 °C, allowing for more specific hybridization at lower temperature [32]. For monovalent ions, 0.5-1M sodium chloride and sodium citrate (SSC) reduce electrostatic repulsion between the two DNA strands and hence enhance hybridization. Decreasing salt concentration applies more stringent conditions, encouraging specific binding to occur. Overnight probing ensures that hybridization is approaching equilibrium and therefore increases specificity [33]. Specificity also increases by increasing washing volume while sensitivity decreases as sample concentration decreases. Secondary structures of target molecules are of significant concern in DNA microarray hybridization experiments, as they affect both sensitivity and specificity [34]. Secondary structure formation may reduce the target binding constant by as much as 105 to 106 times [35]. Several approaches, such as target denaturation, helper oligonucleotides [36] and target fragmentation [34,37,38] have been adopted for RNA based oligonucleotides microarrays to reduce target or probe self folding effects.
Several methods have been described for target DNA labelling. Direct labelling using fluorescently labelled dNTPs during cDNA synthesis is the simplest and quickest way to incorporate label into cDNAs [20]. However, this method has two disadvantages. Firstly, the incorporation efficiency of reverse transcriptase is reduced by the more bulky labelled nucleotides. Secondly, the labelling is sequence dependent as only one modified nucleotide is used. The first drawback can be overcome by using an indirect labelling method, such as incorporating aminoallyl modified nucleotides that are then coupled to NHS fluorescent dye. The need for extra purification steps in this indirect method makes it slightly more expensive and complicated than the direct labelling [16]. Dendrimer end labelling is one of the most efficient methods that has been reported to label oligonulceotides [39-41]. Dendrimers are branched molecules incorporating multiple fluorescent dyes into a single molecule. Briefly, cDNA is synthesized using normal dNTPs using an oligo (dT) primer containing a specific capture sequence at the 5’ end. The resulting unlabelled cDNAs is hybridized to the array. The array is then probed with the dendrimer modified oligonucleotides that are complementary to the 5’ capture sequence. This method shows improved sensitivity, as the signal obtained from 1-2 µg of total RNA is generally equivalent to that obtained with 40-50 µg of total RNA using the direct labelling method [16]. The dendrimer method amplifies signal for low abundance RNA transcripts without affecting the representation of relative abundance for different transcripts. PCR based amplification is another approach that amplifies low abundance RNA transcripts without being sequence dependent. It requires only picogram amounts of RNA as amplification efficiency can be as high as 3 x 1011 fold [42]. An oligo dA tail added by terminal deoxynucleotidyl transferase to the 3’ end of reverse transcribed cDNA provides a priming site. Oligo (dT) primers are then used in subsequent conventional PCR cycles. Although this method is easy, quick and cost effective, it has two potential problems which may cause a failure to preserve the relative RNA abundance for all transcripts: failure to introduce the priming site or the inefficiency of 3’ biased PCR reactions in the case of long (over 500 bp) cDNAs [42,43]. All the previous labelling methods generate a labelled version of the full length transcript.
In this manuscript, we optimise and compare the specificity and sensitivity of the dendrimer and a modified PCR based labelling method. HEK293 cells were separately transfected with 8 plasmids that are identical except for a unique 23 bp sequence (UR). All these URs are closely related with a degree of identity ranging from 35% to 74% (Supplementary information, Table S1). Multiple mismatches are distributed over the entire UR sequences so that each UR can act as a mismatch to the remaining ones (Supplementary information, Fig. ( S2)). Neither a dA tail nor dT primer was used in the modified PCR method in order to preserve RNA transcript relative abundance. Instead, a universal primer pair was used to generate 120 bp short amplicons of equal size for all transcripts. Full length and 330 bases transcripts generated using the Genisphere oligo dT and customized antisense primers respectively Fig. (1) were used to assess the dendrimer technology.
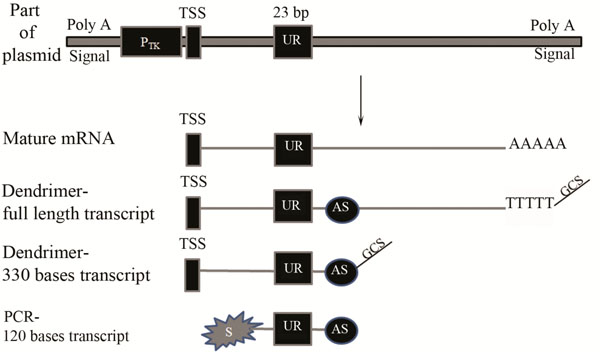
A Schematic diagram of part of the plasmid, transcribed mRNA and cDNA labelled by either the dendrimer technology or the modified PCR based approach.
A unique reporter (UR) sequence of 23 bp was constructed downstream of thymidine kinase promoter (Ptk) and the whole region was flanked by Poly A signal. TSS represents the transcription start site. AS refers to the antisense primer binding site. S represents the Cy3 labelled sense strand used for PCR amplification. The Genisphere capture sequence is represented by GCS.
2. MATERIALS AND METHODS
2.1. Slide Fabrication and Blocking
Corning epoxy coated glass slides were used as the DNA microarray substrate. HPLC purified oligonucleotides were purchased from MWG Eurofins (Germany). Sense and antisense oligonucleotides (Table 1) were designed to capture antisense and sense strands of probes generated by the dendrimer and the modified PCR based labelling methods respectively. Each capture oligonucleotide was diluted in 0.15 M NaH2PO4 spotting buffer (pH 8.5) to a final concentration of 25µM and transferred to a 96 well polypropylene plate (Greiner, UK). Slides were printed using a Scienion SciFlexarrayer S3 piezoelectric arrayer (Germany) according to the manufacturer’s protocol in a dust free environment. The arrayer was designed to produce 16 subarrays, with ten replicates of each UR capture in each subarray. Double deionized water was used to clean the nozzles and to prevent oligonucleotides carry over. Fabricated slides were incubated overnight at ambient temperature in a 70% relative humidity environment in a humidity chamber using a saturated solution of NaNO2. Fabricated slides were pretreated in Coplin jars with blocking solution containing 5 x SSC, 0.1%BSA and 0.1% SDS. Slides were blocked for 90 minutes at 42°C and then washed three times in 0.1 x SSC solution at ambient temperature for 5 minutes each. This was followed by one washing in purified water at ambient temperature for 3 minutes. Slides were then dried by centrifugation at 1,600 x g for 5 minutes.
Antisense and Sense Captures Sequences for Unique Reporters (URs)
| UR | Anti sense Captures (5’-3’) | Sense Captures (5’-3’) |
|---|---|---|
| PMUR 10 | CCTCCACACATTTCGCTCAGACC | GGTCTGAGCGAAATGTGTGGAGG |
| PMUR 11 | GGTCCACACAATCGGCTTAGTCG | CGACTAAGCCGATTGTGTGGACC |
| PMUR 13 | CGTTTACACCGTCCGCTCAATCC | GGATTGAGCGGACGGTGTAAACG |
| PMUR 14 | GCCTCAAACAGTCCGCTAAGTCC | GGACTTAGCGGACTGTTTGAGGC |
| PMUR 15 | CGTCCACACAGTCGTAGCAGTCC | GGACTGCTACGACTGTGTGGACG |
| PMUR 16 | CGTCCACACAGTCCGAGAGGTTG | CAACCTCTCGGACTGTGTGGACG |
| PMUR 17 | CGTCCACTGCCGTCGCTATTATC | GATAATAGCGACGGCAGTGGACG |
| PMUR 18 | GGAGCACAGACGACGATAGGACG | CGTCCTATCGTCGTCTGTGCTCC |
| MMUR 11 | GGTCCACACAACCGGCTTAGTCG | |
| MMUR 14 | GCCTCAAACAGCCCGCTAAGTCC | |
| MMUR 17 | CGTCCACTGCCATCGCTATTATC |
Antisense-oligonucleotides were used to capture sense stands generated by the modified PCR based labelling method. Sense-oligonucleotides were used to capture antisense strands generated by the dendrimer technology. PM and MM refer to perfect and mismatch respectively.
2.2. Transfection, RNA Isolation and mRNA Purification
Eight plasmids were constructed to be identical except for a 23 bp reporter sequence (UR) unique to each plasmid. These UR sequences are closely related with 35-74% of identity to act as mismatches to each other (Supplementary information, Table S1 and Fig. ( S2). UR sequences were constructed downstream of the thymidine kinase promoter (PTK) (Fig. 1). Each UR sequence is considered to be a mismatch to all other URs. Approximately 106 HEK293 cells (LGC Standards, UK; ATCC Number CRL-1573) were transfected with 200 ng of one of the eight plasmids using GeneJuice® Transfection Reagent (Novagen, UK). Cells were grown at 37 °C for 24 hours in an atmosphere that contained 5% CO2 and then harvested. Total RNA was prepared using the miRNeasy mini kit (Qiagen, UK) with on column DNase digestion step according to the manufacturer’s protocol. The mRNA was purified from 4 μg total RNA using Dynabeads mRNA purification kit (Invitrogen, UK) according to manufacturer’s protocol.
2.3. Target Preparation and Hybridization Using the Modified PCR based Labelling Approach
First strand cDNA was synthesized directly from 10 μl Dynabeads bound mRNA suspension (prepared above). Each 20 μl reaction contained 1 μl dNTP (10 mM each), 4 μl 5X First Strand Buffer, 2 μl 0.1 M DTT, 40 units of RNaseOUT and 200 U of SuperScript II reverse transcriptase (Invitrogen, UK). The reaction was incubated at 42 °C for 52 min, heated to 70 °C for 15 min and cooled to 4 °C. For Second strand cDNA synthesis each sample was amplified separately in a 20µl PCR reaction using universal Cy3 labelled sense primer (Cy3- 5’-GCACCTCTTCGGCAAG-3’) and antisense primer (5’-TCTCCGACCGACATCTG-3’) (Fig. 1). Each PCR reaction included 3µl first strand cDNA, 200µM of each dNTP (Promega, UK), 1x PCR buffer, 1.5mM MgCl2, 0.5 unit of HotStarTaq DNA polymerase (Qiagen, UK) and 1µM of each sense and antisense primer (MWG Eurofins, Germany). The Eppendorf MasterCycler epgradient cycler (Eppendorf, UK) protocol had an initial denaturation of 95°C for 15 minutes followed by 30 cycles of 95°C for 30 sec, 50°C for 30 sec, and 68°C for 30 sec. Final extension was carried out at 68°C for 2 minutes. The reaction was terminated by incubation at 4°C. Fluorescently labelled 120 bp PCR products were purified using the QIAquick PCR purification kit (Qiagen, UK) according to manufacturer’s protocol to remove unincorporated nucleotides, DNA polymerase and primers. Purified amplicons were quantified using the ND-1000 spectrophotometer (NanoDrop Technologies, USA). A range of hybridization buffers were prepared to have 1% SDS and 0.1 mg/ml sonicated salmon sperm DNA in common with varying amount of formamide 10%, 20% or 30% and either 5x or 2x SSC. Diluted amplicons were denatured at 95°C for 5 minutes then centrifuged briefly to compensate for evaporation and kept at either room temperature or 53°C until hybridization. Arrays were hybridized overnight in a humidified chamber at 53°C using either the FAST® Frame Cassette (Sigma, UK) or the Hybrislip™ (Grace Bio Labs, UK). Slides were then briefly immersed in Coplin jars containing 2 x SSC and 0.1% SDS washing solution which were pre heated at 53°C. They were then transferred into a new jar containing the same solution and kept for 5 minutes at 53°C. This was followed by washing in 1 x SSC solution twice for 2 minutes each at room temperature. Final washing was carried out for two times in 0.1 x SSC solution for 1 minute each at room temperature. Slides were then dried by centrifugation at 1,600 xg for 5 minutes.
2.4. Target Preparation and Hybridization Using the Dendrimer Labelling Approach
Purified mRNA was eluted from the Dynabeads by heating for 2 min at 80°C with 5 μl of 10 mM Tris HCl (pH 7.5). cDNA was synthesized using the 3DNA Array 50 Expression Array Detection Kit (Genisphere USA) as recommended by the manufacturer. Full length and 330 bases cDNA were produced to investigate the effect of probe length on hybridization efficiency (Fig. 1). The former was synthesized using the Genisphere Cy3-oligo dT based primer (5’-TTCTCGTGTTCCGTTTGTACTCTAAGGTGGAT(17) -3’). The latter was produced by customizing the antisense primer used for PCR amplification above, to include the Genisphere capture sequence at the 5’ end (5’- TTCTCGTGTTCCGTTTGTACTCTAAGGTGGATCTCCGACCGACATCTG -3’). Briefly, 0.5 μg mRNA was reverse transcribed for 2 hours at 42 °C using 15 pmol of either Genisphere or customized primer and 400 units superscript II (Invitrogen, UK). The cDNA was concentrated using a Microcon YM 30 (Millipore, USA) to a final volume of 10 μl. 2.5 μl of the latter was dissolved in 0.5 μl LNA dT blocker, 1.25 μl nuclease free water and 4.25 μl 2x SDS based hybridization buffer (vial 6). The cDNA mix was denatured for 10 min at 80 °C then centrifuged briefly. Arrays were hybridized with this solutiuon overnight in a dark humidified chamber at 53 °C. Slides were then briefly immersed in Coplin jars containing 2 x SSC and 0.2% SDS washing solution which was pre heated at 53°C to remove Hybrislip™. They were then transferred into a new jar containing the same solution and kept for 15 minutes at 53°C. This was followed by one wash cycle in 2 x SSC solution for 15 minutes at room temperature. Final washing was carried out once in 0.2 x SSC solution for 15 minutes at room temperature. Slides were then dried by centrifugation at 1,600 xg for 5 minutes. The slides were then re hybridized with 3DNA Cy3 specific capture reagent (Cy3 labelled dendrimer molecules) for 3 hours at 53°C. The washing and drying steps were repeated again.
2.5. Synthetic 33 bases Target and Hybridization
A 33 bases probe (Cy3- 5’-AGCTTCGACTAAGCCGATTGTGTGGACCGGACA- 3’) was purchased from the MWG eurofins (Germany). This probe was designed to be captured by the antisense strand of PMUR11 capture and hybridization was performed in the same way as for the 120 bases probes.
2.6. Scanning and Quantification of Microarrays
Slides were scanned using a ScanArray Express scanner (Perkin Elmer) with a scan resolution of 10 µm, a photomultiplier tube voltage of 80% and a laser power of 90%. Data acquisition and quantification of spot intensities were carried out using the ProScanArray Express Microarray Analysis software (Perkin Elmer). Adaptive circle was used for spatial segmentation of foreground from background fluorescence intensities for each spot. Mean signal intensity of each spot was calculated by subtracting mean background signal from that of foreground. LOWESS normalization method was applied to negate for spatial and intensity variations within a slide. The mean signal-background for ten spots was used for statistical analysis of the ten replicates.
3. RESULTS AND DISCUSSION
Temperature, salt concentration and formamide percentage are the main variable parameters determining the specificity of DNA microarrays. For short probes of 33 bases hybridized in 10% formamide at 53°C, the cross reactivity of PMUR11 probe to MMUR11 capture was decreased significantly from 76.07% to 53.84% by decreasing salt concentration from 5x to 2xSSC (Fig. 2). The signal obtained for MMUR11 capture was quantified and converted into a relative percentage to that of the PMUR11 capture. Increasing formamide percentage to 20% further decreased cross reactivity to 61.57% and 19.15%, and in 30% formamide to 20.95% and 10.77% using 5xSSC and 2xSSC concentration respectively. Hence using 30% formamide and 2x SSC increase stringency and decrease non specificity dramatically by 65.3%. There are two important considerations in this experiment. The first is that short probes of 33 bases in length require very stringent conditions (30 % formamide and 2xSSC salt concentration) to keep non specificity at a minimal level. Secondly, decreasing salt concentration from 5x to 2xSSC has almost the same effect on specificity as increasing that formamide percentage by 10%. Since an increase of 1% formamide is reported to decreases the Tm by an average value of 0.63 °C (Anderson, 1995) this suggests that either of the above conditions has the effect of increasing stringency equivalent to the hybridization temperature being raised by approximately 6.3 °C, but without the concomitant decrease in sensitivity. Fig. (3A) shows that a high degree of specificity is still achieved, under the same stringent condition of 30% formamide and 2xSSC, for PMUR11 probe against PM and MM captures of UR14 and UR 17 sequences which have less than 5% of cross reactivity in total. The sensitivity in case of 120 bases probes was lost upon hybridization under the same stringent condition. Signals only became apparent under much less stringent conditions by dropping formamide percentage from 30% to 10% and increasing SSC concentration from 2x to 5x (Fig. 3B). These signals were very specific to PMUR11 capture sequences with minimal non specificity (9.64%) to MMUR11 capture sequence and less than 1% to all other captures. Using the same approach, this means that for long probes of 120 bases we had to decrease stringency by altering the salt and formamide concentrations equivalent to lowering hybridization temperature by around 19°C to preserve both the sensitivity and specificity we had for 33 bases probes. This would also suggest that signals lost at high stringency condition are likely to be due to probe length and not related to secondary structure effect, which is stabilized at low temperature. This suggests that longer probe length is one of the stringency parameters that should be taken into consideration to increase specificity of DNA microarray without compromising sensitivity.
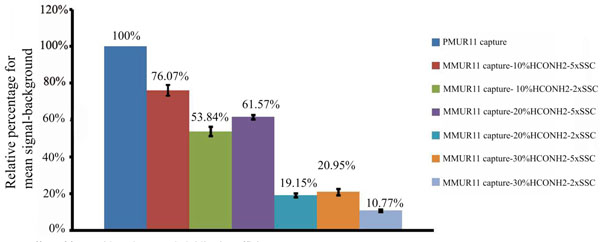
Effect of formamide and SSC on hybridization efficiency.
Relative percentage of mean signal-background for 33 bases probe of PMUR11against PMUR11 and MMUR11 captures using hybridization solutions of variable amounts of formamide 10%, 20% or 30% and either 5x or 2x SSC salt concentration.
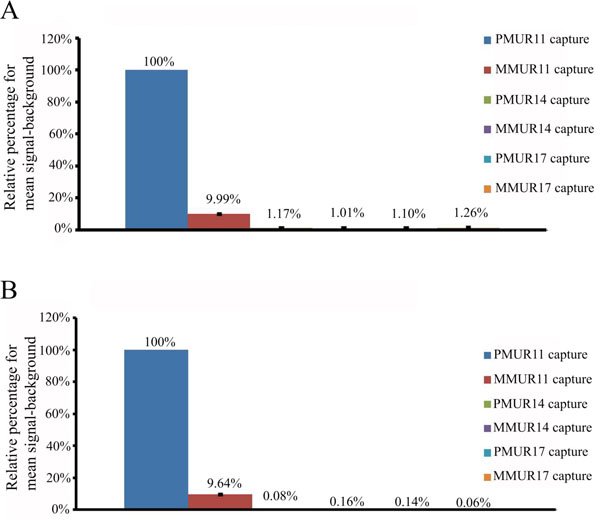
Effect of probe length on hybridization efficiency.
(A) Relative percentage of mean signal-background for 33 bases probe of PMUR11 against PMUR11, MMUR11, PMUR14, MMUR14, PMUR17 and MMUR17 captures using hybridization solution of 30% formamide and 2xSSC salt concentration. (B) Relative percentage of mean signal-background for 120 bases probe of PMUR11 against PMUR11, MMUR11, PMUR14, MMUR14, PMUR17 and MMUR17 captures using hybridization solution of 10% formamide and 5xSSC salt concentration.
The degree of specificity and sensitivity of the probes labelled by either dendrimer technology or the modified PCR approach were compared using the eight different UR capture sequences. These captures are closely related with 35-74% of identity to act as mismatches to each other (Supplementary information, Table S1 and Fig. ( S2). The dendrimer approach was used to detect hybridization of the full length or 330 bases transcripts, while the modified PCR labelling produced 120 bases transcripts (Fig. 1). A high degree of cross reactivity was noticed for all probes using the dendrimer approach. The hybridization temperature suggested by the manufacturer for 30 mer captures is 30-35 °C. However, for the full length transcripts, we found that to enhance the specificity of some probes without compromising sensitivity of any of the probes it was necessary to increase cDNA hybridization temperature to 53 °C. Surprisingly, out of the 8 probes only UR11 and UR15 probes exhibited specific binding (Fig. 4A) with a minimal cross reactivity (0.3-1.5% and 1.2-6.3% respectively) with the percentage of non specific binding for the remaining probes having high values ranging from 2.5% to 333.3% (Supplementary information, Table S3). The absence of signals for arrays hybridized directly with the dendrimer molecules, without using target molecules, indicates that the dendrimer molecules do not cross react with non complementary sequences (data not shown). The specific hybridization observed in case of UR11 and UR15 probes further confirms that the dendrimer molecules bind only to complementary sequences. Although UR11 probe showed a minimal degree of cross reactivity (0.9%) towards UR18 capture, the UR18 probe on the contrary had 205% binding to UR11 capture. This suggests that non specificity may be dependent on probe self folding as well as UR sequence similarities.
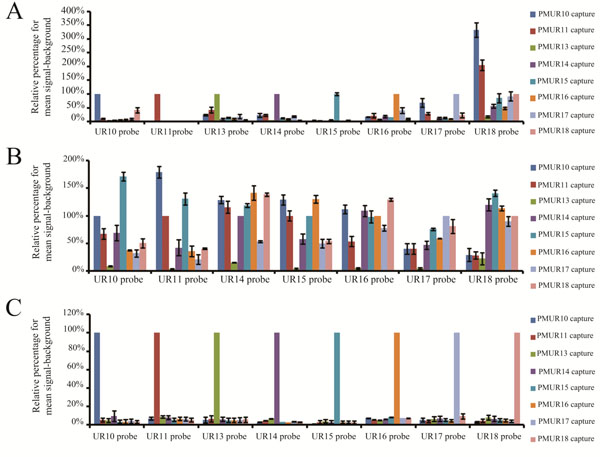
Specificity comparison for full length, 330 bases and 120 bases probes labelled with either the dendrimer technology and the modified PCR based method.
(A) Relative percentage of mean signal-backround for each of the eight full length transcripts against each unique reporter capture sequence. Transcripts were labelled by the dendrimer technology. (B) Relative percentage of mean signal-backround for each of the eight 330 bases transcripts against each unique reporter capture sequence. Transcripts were labelled by the dendrimer technology. (C) Relative percentage of mean signal-backround for each of the eight 120 bases transcripts against each unique reporter capture sequence. Transcripts were labelled by the modified PCR method.
Although increasing the probe length from 33 to 120 bases increases stringency and specificity, the failure of the dendrimer technology to preserve specificity for most of the full length probes could be related to probe secondary structure formation that hinders efficient probe capture hybridization. In an attempt to minimize secondary structure formation, shorter probes were generated for evaluation using the dendrimer approach. The Genisphere oligo dT based primer was replaced by a customized antisense primer to produce cDNA of 330 bases in length (Fig. 1). Shorter transcripts were allowed to hybridize at higher stringency conditions of 57 °C to attempt to obtain a high degree of specificity without compromising sensitivity. None of these transcripts hybridized specifically to its corresponding capture (Fig. (4B) and Supplementary information, Table S4). Unlike for the full length transcripts, UR11 and UR 15 probes of these shorter transcripts have totally lost specificity. One explanation for the loss of specificity of UR11 and UR15 probes in the 330 bases transcripts could be that there is some secondary structure formation that masks the UR sequence which is lost in the full length transcripts allowing exposure of UR sequences for specific capture binding.
One of the limiting factors that we have encountered with the dendrimer approach was the length of the probes. The shortest transcript that we could produce, using a customized antisense primer 41 bases downstream from the 3’ end of the UR sequence, was 330 bases (Fig. 1). The high values of non specificity obtained from the dendrimer approach with either full length or 330 bases transcripts indicate that it could not be used to differentiate between closely related probes and therefore would not be suitable for downstream applications such as determination of differential gene expression. Unlike the dendrimer technology, the modified PCR bases approach can easily generate variable lengths of transcripts. Decreasing probe length to 120 bases by the modified PCR based labelling (Fig. 1) increased the degree of specificity for all the probes towards their corresponding captures (Fig. 4C). Non specific binding was minimum where the highest percentage for cross reactivity reaching 9.6% (Supplementary information, Table S5). The percentage range for non specific binding of the 8 probes was between 1% and 9.6% probably being more representative of background noise than sequence dependence. The high degree of specificity for all 120 bases probes generated by the modified PCR method suggests that there was minimal self folding for these probes. Probe capture pairs were designed to have similar melting temperatures and not to be self folded and the specific hybridization for all probes at the same temperature (53°C) using the PCR based method indicates that this was achieved. It also suggests that the capture design did not contribute to the failure of dendrimer labelling to maintain specificity. In fact, specific hybridization of the full length transcript for UR11 and UR15 probes further supports this. This clearly indicates that the PCR based labelling method is superior to the dendrimer approach in enhancing specificity of the closely related eight UR sequences.
It is clear that the stringent conditions that ensure maximum specificity for short probes of 33 bases are not likely to be as suitable for the same probe sequence in the oligonucleotides of increased length of 120 bases. Sensitivity of these longer probes could only be enhanced, without losing specificity, under less stringent conditions. This suggests that increasing probe length itself represents one of the stringency conditions that can be used to increase specificity. Increasing target length to 120 bases using the modified PCR labelling method showed that closely related probes bind specifically to their corresponding captures with minimal non specificity even under relatively lower stringency conditions. On the other hand, full length or 330 bases transcripts (as used in the dendrimer technology) resulted in a high degree of cross reactivity even under high stringency condition, possibly as a result of secondary structure formation. Our data suggests that reducing probe length increases target sequence exposure to its relevant complementary capture and therefore would enhance hybridization efficiency. These results are in line with other findings on target fragmentation effect [37,38]. Our data does not support Lane’s and coworkers’ findings [44] which suggested that reducing PCR amplicon size had no effect on secondary structure and therefore did not enhance specificity. All the eight PCR amplicons which were approximately 200 bases apart from each others [44] were hybridized at the same condition and this would not be favoured based on our findings. Second, the shortest PCR amplicon that was tested had 162 bp and there was no any further reduction in probe length.
Sensitivity is a key issue for DNA microarrays, especially when samples are limited. We compared two hybridization methodologies using the 120 bp amplicons generated by the modified PCR based method, FAST® Frame (Schleicher & Schuell) and Hybrislip™ (Grace Bio Labs). For the FAST® Frame hybridization cassettes the minimal probe volume that ensured that each subarray remained covered after overnight hybridization was 120 µl. Saturation of arrays generated from spotting 25 µM capture molecule solutions was achieved with 0.5 pmol total probe with the minimal target molecule detectable being 1fmol total per well (Fig. 5A). The detection limit using this system would not be sufficient to detect low abundance transcripts. Therefore, Hybrislip™ were tested as a second option for hybridization. The capability of using 2 µl of probe solution for each subarray made it possible to detect down to 0.01fmol total target molecules and 0.1 pmol of target molecules was sufficient to saturate the capture (Fig. 5B). Decreasing hybridization volume from 120 µl to 2 µl increased sensitivity by 100 fold. In an attempt to further enhance assay sensitivity of the PCR based labelling method, the effect of post denaturation temperature was investigated, using room and hybridization temperatures. It was noticed that denatured probes kept at room temperature, prior to hybridization, produced higher mean signal-background than those kept at the hybridization temperature. This might be due to rapid reannealing of denatured strands kept at hybridization temperature compared to those kept at room temperature. Therefore denatured samples were kept at room temperature to increase sensitivity of the assay.
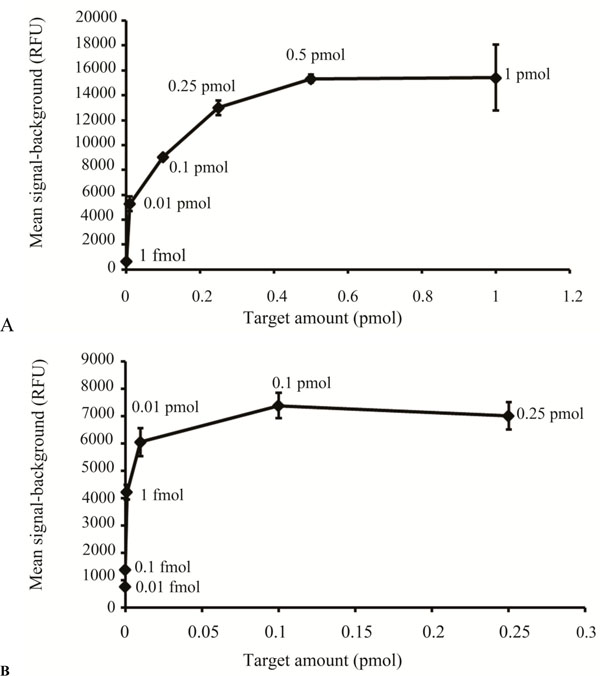
Sensitivity comparison between the FAST® Frame Cassette and the Hybrislip™.
(A) Different amounts (pmoles) of the same target, generated by the modified PCR method are plotted against the mean signal-background (RFU) for hybridization carried out in the FAST® Frame hybridization chamber. (B) Different amounts (pmoles) of the same target, generated by the modified PCR method, are plotted against the mean signal-background (RFU) for hybridization carried out using the Hybrislip™.
4. CONCLUSIONS
The manuscript highlights many of the factors affecting sensitivity and specificity of DNA microarray. Target length is one of these critical parameters that should be carefully considered. It is crucial to choose the optimum length that would ensure maximum specificity of the DNA microarray without compromising sensitivity. In addition to probe length, other factors such as hybridization chambers, formamide percentage, salt concentration, and temperature have also been investigated.
The manuscript also indicates the importance of choosing the appropriate labelling method that would not interfere with either sensitivity or specificity. The ability of the modified PCR based approach to amplify low abundant transcripts using primers of choice leads to generation of wide range of amplicon sizes. This indeed gives the modified PCR labelling method more advantages and flexibility over the dendrimer approach in managing specificity and sensitivity of the DNA microarray hybridization.
CONFLICTS OF INTEREST
The authors confirm that this article content has no conflicts of interest.
ACKNOWLEDGEMENT OF FUNDING
This study was funded by the EPSRC
SUPPLEMENTARY MATERIAL
This article also contain supplementary data and it can be viewed at Download Supplementary Material
ACKNOWLEDGEMENTS
The author would like to thank Prof. A. Pitt, Prof. W. Kolch, Dr. E. McKenna and Dr. S. Gannon for their valuable discussion and for reading the manuscript. This project was financially supported by the EPSRC grant.

