All published articles of this journal are available on ScienceDirect.

Biophysical Mechanisms of the Neutralization of Endotoxins by Lipopolyamines
Authors Info & Affiliations
Abstract
Endotoxins (lipopolysaccharides, LPS) are one of the strongest immunostimulators in nature, responsible for beneficial effects at low, and pathophysiological effects at high concentrations, the latter frequently leading to sepsis and septic shock associated with high mortality in critical care settings. There are no drugs specifically targeting the pathophysiology of sepsis, and new therapeutic agents are therefore urgently needed. The lipopolyamines are a novel class of small molecules designed to sequester and neutralize LPS. To understand the mechanisms underlying the binding and neutralization of LPS toxicity, we have performed detailed biophysical analyses of the interactions of LPS with candidate lipopolyamines which differ in their potencies of LPS neutralization. We examined gel-to-liquid crystalline phase behavior of LPS and of its supramolecular aggregate structures in the absence and presence of lipopolyamines, the ability of such compounds to incorporate into different membrane systems, and the thermodynamics of the LPS:lipopolyamine binding. We have found that the mechanisms which govern the inactivation process of LPS obey similar rules as found for other active endotoxin neutralizers such as certain antimicrobial peptides.
INTRODUCTION
Infectious diseases continue to be a major cause of preventable mortality worldwide. Alarming increases in the incidence of multi-drug resistance in the face of a dwindling pipeline of new antibiotics is perhaps a scenario that Fleming could not have contemplated. Indeed, the incidence of sepsis continues to increase worldwide which may, in part, be attributable to compromised efficacy of existing antibiotics. Inadequate antimicrobial chemotherapy may lead to a release of bacterial pathogenicity factors such as lipopolysaccharides (LPS, endotoxins) and lipoproteins (LP) [1-3]. Moreover, in some common viral infections such as influenza, it is known that cases with severe outcome are in most cases due to a superinfection, i.e., the initial viral infection is followed by a bacterial infection, an example being the ‘Spanish flu’ in the 1920’s, in which millions of victims did not die from primary infection with influenza H1N1 [4], but from the subsequent bacterial infections, often leading to sepsis [5-7].
Two recent approaches in addressing this urgent, unmet medical need are the use of anti-LPS or anti-LP compounds, which have bind to, and neutralize these toxins so that they are no longer able to interact with upstream binding proteins such as lipopolysaccharide-binding protein (LBP), CD14, and the TLR4/MD2 complex, thereby preventing the activation of proinflammatory responses in target cells such as monocytes and macrophages. Notable examples of such sequestering compounds are the antimicrobial peptides termed synthetic anti-LPS peptides (SALP) [8,9] and the lipopolyamines (LPA) [10-15]. The biophysical mechanisms of the endotoxin:SALP interaction was published in recent reports [8,16]. In these reports it was found that the driving force of the interaction is the Coulomb attraction between the cationic groups of the SALP and the negative charges of the endotoxin with a subsequent intercalation of the hydrophobic moiety of the SALP into the lipid A moiety of LPS, inducing a change of the aggregate structure of the latter into a multilamellar assembly [8,9,16]. For the LPA:endotoxin interaction, such data are lacking. A detailed understanding of the biophysical correlates of mechanisms underlying the complexation of LPA with LPS, and subsequent neutralization of endotoxicity is of particular relevance to further optimize efficacy. Hence, the main focus of this study was to obtain a detailed characterization of the neutralization mechanisms of LPS by LPA. The data show similar effects as published for SALP, from which the general structural correlates for effective anti-endotoxin action can be deduced.
MATERIALS AND METHODS
Lipids and Lipopolysaccharides
Lipopolysaccharides from Salmonella minnesota rough Re and Ra, strains R595 and R60, respectively, were extracted from the bacteria grown at 37 °C by the phenol:chloroform: petrol ether (PCP) method, purified, and used in the natural salt form [17]. The purity was examined by MALDI-TOF mass spectrometry, and LPS samples were only used when the chemical structure in particular of the lipid A part consisted of a diglucosamine, to which six acyl chains in amide and ester-linkage at positions 2 and 2’, and 3 and 3’, respectively were bound and which were phosphorylated at positions 1 and 4’, according to the known structure of lipid A [18,19].
For all experiments except the SAXS measurememts (see below), as endotoxin LPS Ra was taken since this compound is within the heterogeneous LPS wild-type the bioactive compound [16,19].
The phospholipids phosphatidylcholine (PC) and phosphatidylserine (PS) were purchased from Avanti Polar Lipids (Alabaster, Al, USA). The lipopolyamines YAN-673, DS-347 and DS-176 (Schemes 1, 2 and 3) were synthesized starting from DS-96 [14] as follows.
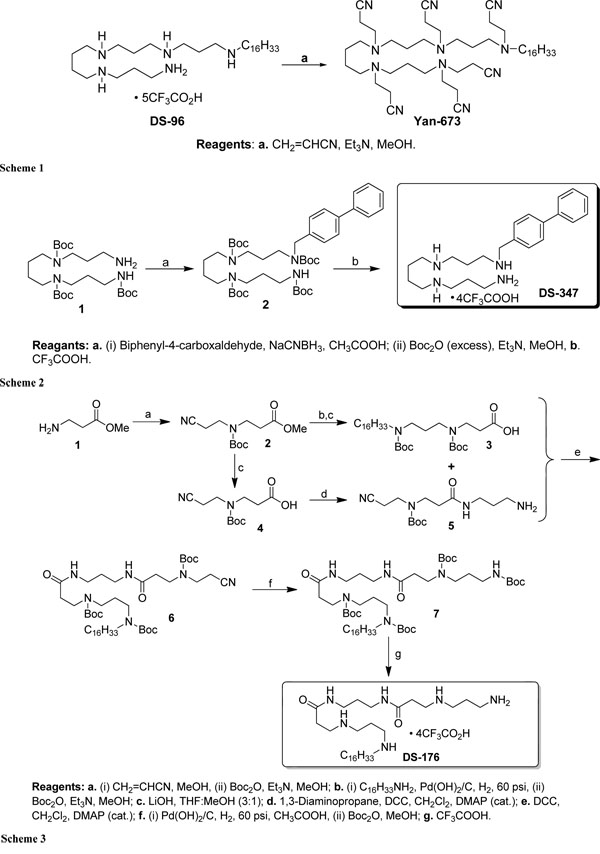
Schematic presentation of the synthesis of the lipopolyamines.
Synthesis of Yan-673 (Scheme 1): To a solution of DS-96 (0.3 g, 0.285 mmol) in anhydrous methanol (30 mL) at room temperature was added acrylonitrile (0.188 mL, 2.85 mmol), triethylamine (0.198 mL, 1.43 mmol) and the mixture was stirred at room temperature for 8 h. The resulting solution was concentrated in vacuum, and purified by flash column chromatography (MeOH: DCM = 10:90) to give the colorless oil YAN-673 (0.2 g, 87.7%). 1H NMR (500 MHz, CDCl3) δ 2.85 (t, J = 6.7 Hz, 4H), 2.80 – 2.72 (m, 8H), 2.62 (t, J = 6.9 Hz, 2H), 2.56 – 2.40 (m, 27H), 1.65 – 1.54 (m, 6H), 1.51 – 1.37 (m, 6H), 1.32 – 1.21 (m, 27H), 0.87 (t, J = 6.9 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 119.5, 119.4, 119.3, 118.8, 53.8, 53.63, 53.61, 51.6, 51.5, 51.1, 50.9, 49.6, 49.5, 49.4, 49.3, 49.2, 31.9, 29.7, 29.68, 29.67, 29.6, 29.4, 27.4, 27.1, 25.4, 25.1, 24.9, 22.7, 16.9, 16.7, 16.6, 16.5, 16.3, 14.1. MS (ESI) calculated for C47H83N11, m/z 801.6833, found 802.7576 (M + H) +.
Synthesis of DS-347 (Scheme 2): To a solution of tri-Boc spermine 1 (0.65 g, 1.29 mmol), which was synthesized from spermine using a reported procedure [20], was added acetic acid (0.1 mL) in anhydrous methanol (30 mL) followed by the addition of biphenyl-4-carboxaldehyde (0.23 g, 1.26 mmol), and sodium cyanoborohydride (0.120 g, 1.91 mmol). The colorless solution was stirred at room temperature for 24 h. The solution was concentrated under reduced pressure, and the residue was extracted with dichloromethane (3 × 50 mL). The combined organic layers were washed with brine and dried over anhydrous Na2SO4. After removal of the solvent under high vacuum, the crude biphenyl amino derivative was dissolved in methanol (20 mL) to which a solution of di-tert-butyl dicarbonate (1.3 g, 5.96 mmol) in methanol (5 mL) was added. Removal of solvent and purification of the crude product by column chromatography (Hexane:EtOAc = 25:75) provided the compound 2 as a colorless oil. The compound 2 (0.5 g, 0.651 mmol) was dissolved in anhydrous trifluoroacetic acid (20 mL) and stirred at room temperature for 30 min. The solvent was removed by purging nitrogen and the residue was washed with diethyl ether to obtain the white solid DS-347 as tetra trifluoroacetate salt (0.461 g, 86%). 1H NMR (400 MHz, DMSO) δ 9.22 (s, 1H), 8.88 (bs, J = 6.4, 3.4, 1.8 Hz, 2H), 7.97 (bs, J = 0.7 Hz, 2H), 7.75 (d, J = 8.0 Hz, 2H), 7.69 (d, J = 7.8 Hz, 2H), 7.58 (d, J = 8.0 Hz, 2H), 7.49 (t, J = 7.6 Hz, 2H), 7.40 (t, J = 7.2 Hz, 1H), 4.21 (s, 2H), 3.14 – 2.78 (m, 12H), 2.09 – 1.96 (m, 2H), 1.95 – 1.82 (m, 2H), 1.63 (s, 4H). 13C NMR (101 MHz, DMSO) δ 141.3, 139.8, 131.4, 131.0, 129.5, 128.3, 127.4, 127.2, 50.2, 46.6, 44.4, 44.3, 44.2, 36.7, 24.3, 23.1, 22.9. MS (ESI) calculated for C23H36N4, m/z 368.2940, found 369.3060 (M + H) +.
Synthesis of DS-176 (Scheme 3): To a solution of β-alanine methyl ester hydrochloride (3.5 g, 25.07 mmol) in anhydrous methanol (30 mL) was added acrylonitrile (1.65 mL, 33.9 mmol) and the mixture stirred at room temperature for 15 h. After removal of solvent under high vacuum, the crude nitrile derivative was dissolved in 30 mL of methanol followed by the addition of di-tert-butyl dicarbonate (10.94 g, 50.14 mmol). The resulting solution was stirred for 8 h at room temperature, concentrated in vacuum, and purified by flash column chromatography (EtOAc:Hexane = 14:86) to give a colorless oil 2. The mono-nitrile derivative 2 (1.9 g, 7.42 mmol) and hexadecylamine (5.4 g, 22.26 mmol) in methanol (25 mL) was hydrogenated over Pd(OH)2/C s catalyst at 2.76 bar pressure for 12 h. The catalyst was filtered on celite and the solvent was removed under reduced pressure, which was used for the next step without purification. To this crude compound in methanol (20 mL) were added di-tert-butyl dicarbonate (7.00 g, 32.00 mmol) and triethylamine (1.04 mL, 7.42 mmol). The resulting solution was stirred for 2 h at room temperature. The solvent was then removed under reduced pressure and purified by flash column chromatography (EtOAc: Hexane = 10:90) to give the methyl ester derivative (3.78 g, 6.46 mmol) as a viscous oil which was dissolved in THF (10 mL) followed by the addition of a solution of lithium hydroxide (0.47 g, 19.38 mmol) in water (20 mL). The resulting solution was stirred at room temperature for 24 h. The reaction mixture was extracted with chloroform and the combined extracts were washed with water, brine, and dried over sodium sulfate. After removal of solvent under vacuum, the residue was purified by flash column chromatography (MeOH: DCM = 10:90) to afford the compound 3 as colorless oil (3.5 g). In a similar way the ester derivative 2 was hydrolysed with lithium hydroxide to give the compound 4. The compound 4 (4.76 g, 19.6 mmol) was then coupled with 1,3-diaminopropane (8.21 mL, 58.8 mmol) in anhydrous dichloromethane using dicyclohexylcarbodiimide (4.1 g, 19.8 mmol) (DCC) and a catalytic amount of 4-(Dimethylamino)pyridine (DMAP) to give the crude amine 5. The crude amine 5 (1.6 g, 5.36 mmol) was finally coupled with the compound 3 (1.82 g, 3.2 mmol) in anhydrous dichloromethane using DCC (0.65 g, 3.15 mmol) and a catalytic amount of DMAP to give the nitrile derivative 6. The nitrile derivative 6 (1.46 g, 1.72 mmol) was hydrogenated over Pd(OH)2/C as catalyst at 2.76 bar pressure in glacial acetic acid (20 mL) for 12 h. Acetic acid was removed under reduced pressure after filtration of the catalyst on celite followed by the addition of di-tert-butyl dicarbonate (2.46 g, 11.3 mmol) in methanol (15 mL). After removal of solvent under vacuum, the residue was purified by flash column chromatography (MeOH:DCM = 4:96) to yield the protected amine derivative 7 as a colorless oil. The compound 7 (1.16 g, 1.22 mmol) was dissolved in anhydrous trifluoroacetic acid (20 mL) and stirred at room temperature for 30 min. The solvent was removed by purging nitrogen and the residue was washed with diethyl ether to obtain the white solid DS-176 as tetra trifluoroacetate salt (1.0 g, 81.3%). 1H NMR (500 MHz, DMSO) δ 8.86 – 8.62 (m, 6H), 8.18 (t, J = 5.4 Hz, 2H), 7.95 (s, 3H), 3.16 – 3.05 (m, 8H), 2.99 (s, 6H), 2.88 (s, 4H), 1.98 – 1.85 (m, 4H), 1.60 – 1.51 (m, 4H), 1.32 – 1.19 (m, 26H), 0.85 (t, J = 6.9 Hz, 3H). 13C NMR (126 MHz, DMSO) δ 168.8, 46.7, 44.0, 43.9, 43.8, 43.0, 36.4, 36.1, 31.3, 30.9, 30.8, 29.01, 29.0, 28.90, 28.88, 28.8, 28.7, 28.5, 25.8, 25.4, 23.6, 22.2, 22.1, 13.9. MS (ESI) calculated for C31H66N6O2, m/z 554.5247, found 555.4236 (M + H) +.
Stimulation of human mononuclear cells by LPS: The stimulation of human mononuclear cells was performed as described previously [21,22]. Briefly, MNC were isolated from heparinized blood of healthy donors. The cells were resuspended in medium (RPMI 1640) at 5x106cells/ml. For stimulation, 200 µl MNC (1x106cells) were transferred into each well of a 96-well culture plate. LPS R60 and the LPS:lipopolyamine mixtures were preincubated for 30 min at 37°C, and added to the cultures at 20 µl per well. The cultures were incubated for 4 h at 37 °C under 5 % CO2. Supernatants were collected after centrifugation of the culture plates for 10 min at 400xg and stored at –20 °C. TNFα concentrations were quantified by a sandwich ELISA using a monoclonal antibody against TNF (clone 6b from Intex AG, Switzerland).
Fluorescence resonance energy transfer spectroscopy (FRET): The ability of the LPA to intercalate into phospholipid liposomes made from phosphatidylcholine (PC) or phosphatidylserine (PS) or into LPS R60 aggregates was investigated by FRET as described earlier [16]. Briefly, phospholipid liposomes or LPS R60 were doubly labelled with the fluorescent phospholipid dyes 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-(7-nitro-2-1,3-benzoxadiazol-4-yl), ammonium salt (NBD-PE) and N-(lissamine rhodamine B sulfonyl)-phosphatidylethanolamine (Rh-PE) (Molecular Probes). Intercalation of unlabelled molecules into the doubly labelled liposomes leads to probe dilution and with that to a lower FRET efficiency: the emission intensity of the donor ID increases and that of the acceptor IA decreases (for clarity, only the quotient of the donor and acceptor emission intensity is shown here). In all experiments, aliquots of the LPA (100 μl of 100 μM) were added to doubly labelled phospholipid liposomes or LPS R60 aggregates (900 μl of 10 μM) at 50 s after equilibration. NBD-PE was excited at 470 nm and the donor and acceptor fluorescence intensities were monitored at 531 and 593 nm, respectively, and the fluorescence signal ID/IA was recorded for further 250 s.
Isothermal Titration Calorimetry (ITC): Microcalorimetric measurements of the binding of LPA to endotoxins were performed on a MCS isothermal titration calorimeter (Microcal Inc.) at 37 °C as described previously [23] LPS R60 (0.05 mM) was dispersed into the microcalorimetric cell (volume 1.3 ml) and the LPA dispersions (1 mM) were filled into the syringe compartment (volume 100 µl). After temperature equilibration, the LPA were titrated in 3 µl portions every 5 min into the lipid-containing cell under constant stirring, and the heat of interaction after each injection measured by the ITC instrument was plotted versus time.
X-ray scattering: For the determination of the aggregate structures of LPS, deep rough mutant LPS R595 was taken, since this also highly bioactive compound provides better resolved spectra than compounds with longer sugar chains [21]. Scattering patterns were determined in the absence and presence of the LPA using small-angle X-ray scattering (SAXS). Due to the amphiphilic nature of the LPA, they may aggregate by themselves and thus were measured also in uncomplexed form. SAXS measurements were performed at the European Molecular Biology Laboratory (EMBL) outstation at the Hamburg synchrotron radiation facility HASYLAB using the double-focusing monochromator-mirror camera X33 [24]. Scattering patterns in the range of the scattering vector 0.001 < s < 0.08 Å-1 (s = 2 sin θ/λ, 2θ scattering angle and λ the wavelength = 0.15 nm) were recorded at 20, 40, 60, and 80 °C with exposure times of 1 min using an image plate detector with online readout (MAR345, MarResearch, Norderstedt/Germany). The s-axis was calibrated with Ag-Behenate with a periodicity of 5.84 nm. The scattering patterns were evaluated as described previously[24] assigning the spacing ratios of the main scattering maxima to defined three-dimensional structures. The lamellar and cubic structures are most relevant in the present study.
Fourier-transform infrared spectroscopy (FTIR) FTIR was used to determine the gel to liquid crystalline phase transition of the hydrocarbon chains of LPS R60 by evaluating the peak position of the symmetric stretching vibration νs(CH2) in the wave number range 2850 to 2853 cm-1. The infrared spectroscopic measurements were performed on an IFS-55 spectrometer (Bruker). LPS and LPS:LPA samples, dispersed in 20 mM Hepes buffer, pH 7.0, were placed in a CaF2 cuvette between 12.5 µm teflon spacers. The temperature-scans were performed automatically between 10 and 70°C with a heating-rate of 0.6 °C/min. Every 3°C, 200 interferograms were accumulated, apodized, Fourier transformed, and converted to absorbance spectra.
Differential Scanning Calorimetry (DSC):Calorimetry measurements were performed with a VP-DSC calorimeters (MicroCal, Inc., Northampton, MA, USA) at a heating and cooling rate of 1 K·min-1 as described by us [25,26]. The DSC samples were prepared by dispersing a known ratio of LPS R60 to the polymer in 10 mM phosphate-buffered saline (PBS), 10 mM phosphate, 138 mM NaCl, 2.7 mM KCl) buffer at pH 7.4. The samples were hydrated in the liquid crystalline phase by vortexing. The measurements were performed in the temperature interval from 5 °C to 95 °C. In the Figures, only the temperature range at which phase transitions were observed is shown. Five consecutive heating and cooling scans checked the reproducibility of the DSC experiments of each sample. The accuracy of the DSC experiments was ΔT = 0.1 °C for the main phase transition temperatures and ΔH = 1 kJ·mol-1 for the main phase transition enthalpy. The DSC data were analyzed using the Origin software. The phase transition enthalpy was obtained by integrating the area under the heat capacity curve.
RESULTS
Lipopolyamines Syntheses
In the Schemes 1-3, the syntheses of the LPAs DS-96 and Yan-673 (Scheme 1), DS-347 (Scheme 2), and DS-176 (Scheme 3) are shown. As can be seen, extensive Michael addition of DS96 yielded the analogue Yan673, in which all the amines are peralkylated (Scheme 1). Furthermore, in compound DS176 (Scheme 3) two of the internal secondary amines are replaced by amides,
For an understanding of the following results, it should be noted that all LPAs consists of two regions, one very apolar region and one with polar characteristics, which is important to know on the basis of the amphiphilic property of the lipid A part of LPS. However, in contrast to SALP the lipopolyamines do not bear free positive charges.
Inhibition of LPS-induced Cytokine Induction:
The production of proinflammatory cytokines such as tumor necrosis factor-α (TNF-α) is an appropriate measure for an inflammation reaction induced by immune stimulants such as LPS R60. Therefore, the first set of experiments was performed to test the ability of the LPA to inhibit the LPS-induced inflammation reaction by measuring the secretion of TNF-α in human mononuclear cells in the presence and absence of different concentrations of LPA (Fig. 1). As can be seen (Fig. 1, top), compounds DS-96 and DS-347 inhibit the cytokine production in a dose-dependent manner; at a [LPA]:[LPS] weight ratio of 100, the activity of LPS is almost completely abrogated. In contrast to this behaviour, the lipopolyamines DS-176 and Yan-673 (Fig. 1, bottom) do not inhibit the LPS-Induced TNF-α production, the latter even enhancing it at the two lower LPS concentrations.
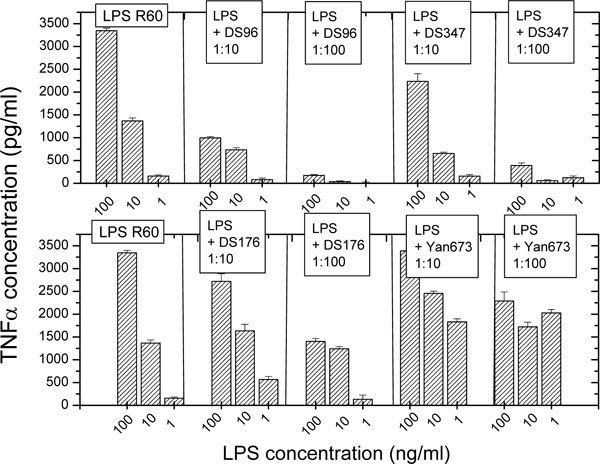
Tumor-necrosis factor-α secretion by human mononuclear cells induced by LPS (from Salmonella minnesota strain R60) in the absence and presence of the lipopolyamines. Error bars (s.d.) were computed from quadruplicate samples.
Gel to Liquid Crystalline Phase Behaviour of LPS
Enterobacterial LPS are known to exhibit a gel to liquid crystalline of the hydrocarbon chains at a temperature Tm = 30 to 36 °C according to the sugar chain length for the different rough mutant LPS [27,28]. The influence of the LPA on the behaviour of LPS R60 was monitored by Fourier-transform infrared spectroscopy (FTIR) by evaluating the peak position of the symmetric stretching vibrational band around 2850 to 2853 cm-1 and by differential scanning calorimetry (DSC) by measuring the melting heat of the hydrocarbon chains at Tm .
Data of FTIR experiments are presented in Fig. (2) for LPS R60 in the absence and presence of DS176 (A) and DS347 (B). As can be seen, pure LPS shows a melting of the hydrocarbon chains at Tm= 36 °C, which decreases in the presence of the LPAs, with DS176 exhibiting a quite moderate decrease to 28 °C at [LPS]:[DS176] 1:2 (weight%), whereas DS347 causes at the highest concentration a complete disappearance of the phase transition. For both compounds, the presence of the LPAs leads to an increase in fluidity (increase in the wavenumber values), which, however, is much higher for DS347 than for compound DS176. The results for compounds DS96 and Yan673 (not shown) are indicative for an only slight decrease in the Tm of LPS, but a decrease in the wavenumbers, whereas Yan673 behaves similar to compound DS347, i.e., a strong increase in the wavenumber values in the entire temperature range. Thus, there is apparently no correlation of these data to their biological action as shown in Fig. (1).
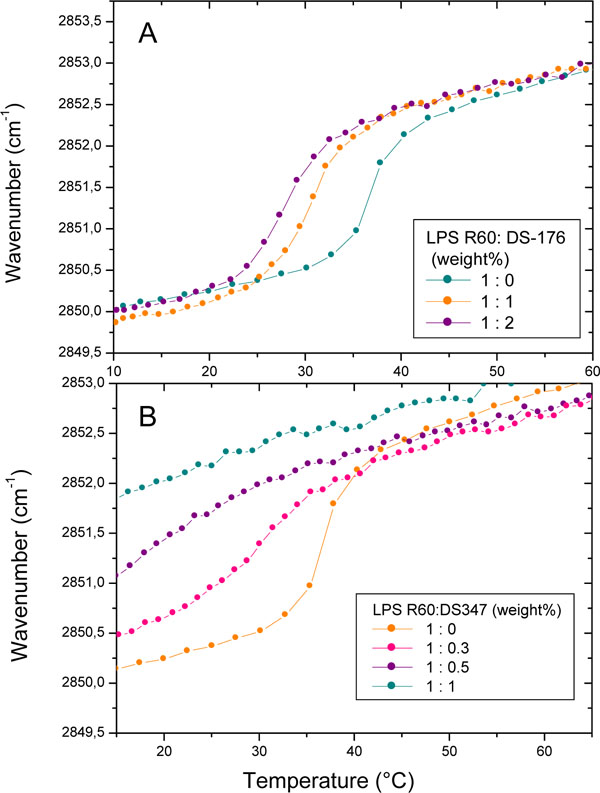
Gel-to-liquid crystalline phase transition of the hydrocarbon chains of LPS R60 at different concentrations of DS176 (A) and DS347 (B) by Fourier-transform infrared spectroscopy. The peak position of the symmetric vibration of the methylene groups νs(CH2) is plotted versus temperature. In the gel phase, the wavenumber values lie at 2850 cm-1, in the liquid crystalline phase they are shifted to 2852.5 to 2853.0 cm-1.
Figs (3A and B) summarize the results from the thermotropic properties of LPS R60 with the polymers DS176 and DS347 from DSC experiments. Various heat capacity curves are shown from the pure lipid up to a LPS:LPA molar ratio of 1:1. The pure lipid LPS R60 shows a gel to liquid crystalline phase transition at ca. 32-33 °C, accompanied by an endothermic phase transition of ca. +20 kJ/mol. The presence of the various polymers exerts differential effects on the thermotropic lipid properties. The presence of small amounts of DS176 reduces the main phase transition temperature of LPS by about 1.5-2 °C. Increasing the amount of DS176 up to a LPS R60: DS176 1:1 molar ratio has no major impact on the phase transition temperature. However, the analysis of the phase transition enthalpy shows that it increases from ca. 20 kJ/mol for the pure lipid up to 29 kJ/mol for the 1:1 mixture. A comparison of the results obtained in the presence of DS347 shows a completely different picture. With increasing amount of the lipopolyamine, the maximum of the phase transition temperature is reduced. Furthermore, it can also be seen that the phase transition enthalpy decreases with increasing amount of polymer. A strong decrease in the phase transition enthalpy is observed at a LPS R60:DS347 molar ratio of 1:0.4 to 1:0.5. Above a molar ratio larger than 1:0.5, no phase transition is observed in the temperature range between 5-50 °C. Even when the samples are stored for 48 h at 4°C, there is no phase transition observed.
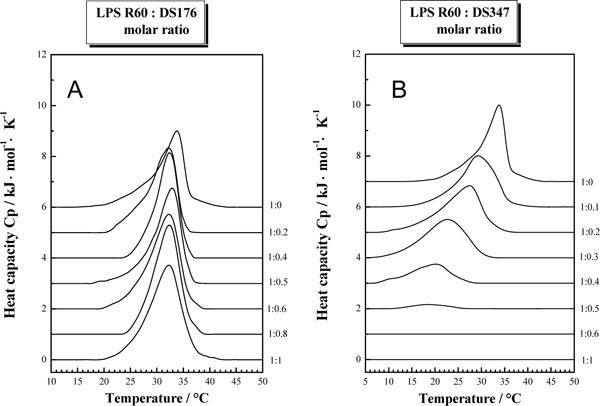
Gel-to-liquid crystalline phase transition of the hydrocarbon chains of LPS R60 at different concentrations of DS176 (A) and DS347 (B) by differential scanning calorimetry. The heat capacity Cp is plotted versus temperature showing the enthalpy change at the phase transition temperature.
Aggregate Structures of LPS:
It has been shown that the aggregate structure of LPS is a determinant of its ability to induce cytokine production in human immune cells [21,28] For unilamellar/cubic aggregate structures, the activity is high, whereas multilamellar structures are characteristic for the absence of bioactivity.
The aggregate structure of LPS was monitored by using small-angle X-ray scattering (SAXS) with synchrotron radiation in the absence and presence of the LPAs. As LPS that from S. minnesota deep rough mutant R595 was taken, since this compound with its short sugar chain gives better resolved SAXS patterns than compounds with long sugar chains. SAXS patterns are shown for LPS R595 in the presence of DS176 (Fig. 4A) and of DS347 (Fig. 4B). For the LPS:DS176 system, SAXS patterns are indicative of only one broad intensity distribution between 0.1 and 0.35/nm, which corresponds to the existence of a LPS bilayer system, e.g, for a unilamellar structure which is frequently found for this endotoxin. Therefore, the addition of compound DS176 does not change the LPS aggregate structure significantly. In the presence of DS347, the patterns exhibit reflections at equidistant ratios. Thus, for example, at 40 °C there are 4 reflections with a main peak at 5.49 nm and further reflections at 2.72 nm = 5.49/2 nm, 1.81 nm = 5.49/3 nm, and 1.35 nm = 5.49/4 nm, which are clearly indicative for the existence of a multilamellar structure of the LPS assembly.
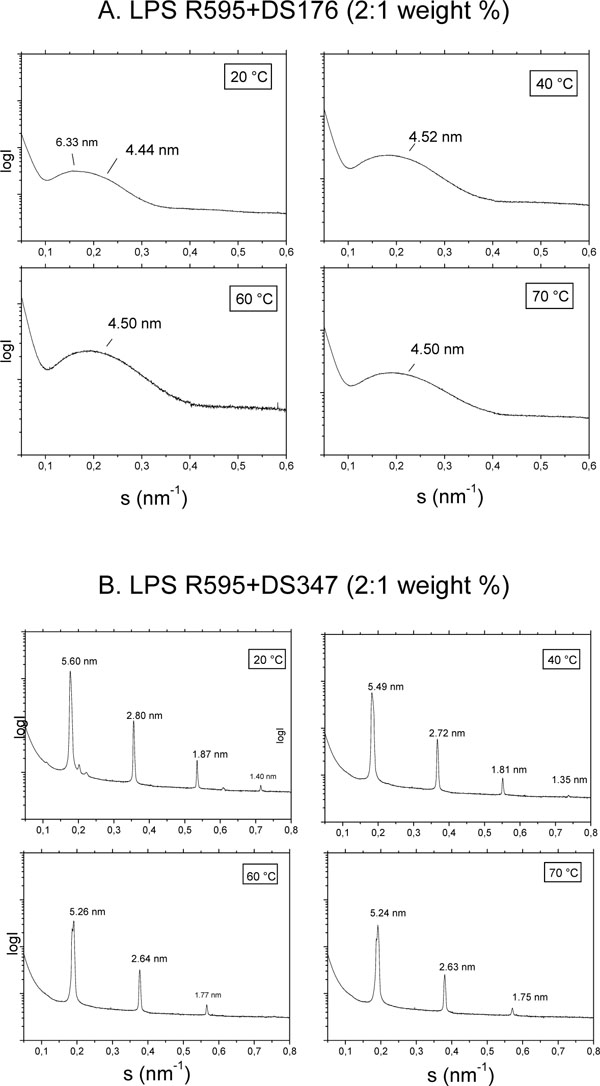
Aggregate structures of LPS R595 in the presence of DS176 (A) and DS347 (B) in small-angle X-ray scattering (SAXS) experiments with synchrotron radiation. The logarithm of the scattering intensity (logI) is plotted versus the scattering vector, s (=1/d, d, spacings of the reflections).
The results for compound DS96 leads similar as for DS347 to the occurrence of reflections typical for a multilamellar phase, whereas for Yan673 a complex superposition of various reflections are indicative of a non-lamellar aggregate structure (not shown).
Incorporation of Lipopolyamines Into lipid Aggregates:
Fluorescence resonance energy transfer (FRET) spectroscopy was applied to detect the possible incorporation of the LPAs into phospholipid molecules characteristic for immune cells such as phosphatidylcholine (PC) and phosphatidylserine (PS) as well as into LPS R60. Data are presented in Figs. (5) for LPS, PC, and PS (from top to bottom).
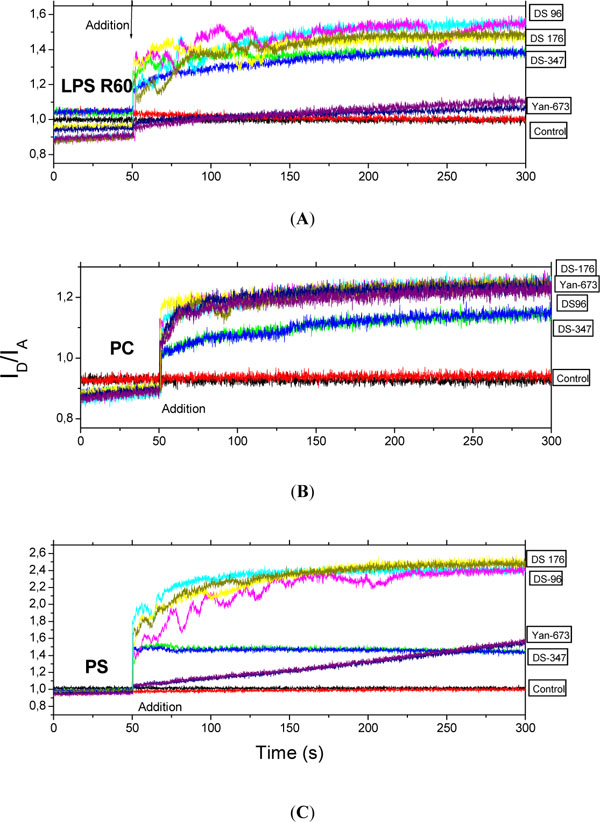
Intercalation of the lipopolyamines into LPS aggregates (A), phosphatidylcholine liposomes (B), and phosphatidylserine liposomes (C) in Förster resonance energy transfer spectroscopic (FRET) experiments. As sensitive measure of incorporation the ratio of donor to acceptor intensity ID/IA is plotted versus time. The LPAs were added to the lipid aggregates after 50 s. Data were generated in two independent experiments (two coloured lines are shown for each compound).
For LPS except for Yan673 all lipopolyamines incorporate similarly into the endotoxin aggregate. For PC, all four investigated LPAs exhibit clearly more or less the same tendency to incorporate into this phospholipid. In contrast, the intercalation of the LPAs is different for PS showing a strong effect for DS176 and DS96, and a much lesser effect for DS347, whereas for Yan673 there is a slowly increasing incorporation which has not finished at the end of the observation time.
From these results it can be concluded that there is apparently no direct correlation with the biological inhibition of the LPS stimulation by the single compounds.
Binding Enthalpy and Stoichiometry of LPS:LPA Mixtures:
To study the kind of binding and its stoichiometry of the LPAs to LPS R60 dispersions, isothermal titration calorimetry (ITC) was applied. In Figs. (6), the results for the two most active compounds DS96 (A) and DS347 (B) are shown: In both cases the interaction consists of only exothermic processes which run into saturation with a sigmoidal shape characteristic for a one target model. The stoichiometry – half saturation values - lies for both compounds around [LPA]:[LPS] = 0.7 to 0.75 molar. This means that 7 to 7.5 LPA molecules are sufficient for the saturation of 10 LPS molecules.
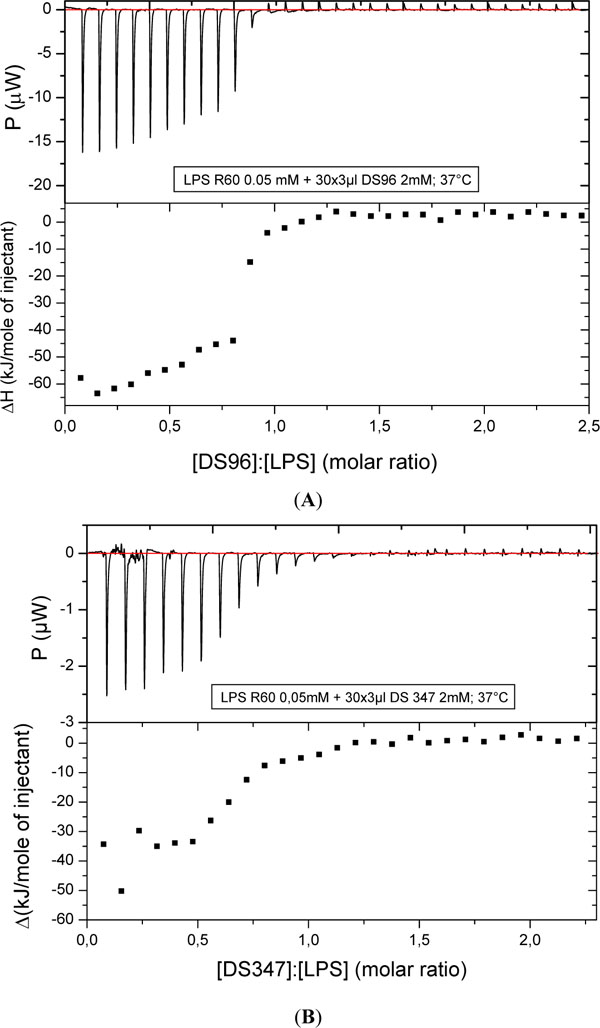
Titration of DS96 (A) and DS347 (B) to LPS R60 and enthalpy change versus [LPA]:[LPS] molar ratio with isothermal titration calorimetry (ITC). The LPA solution (1 mM) was titrated in 3 µl portions to the LPS dispersion (0.05 mM), and the resulting enthalpy change was recorded.
DISCUSSION
We have performed biophysical analyses of the interactions of lipopolyamines with LPS with a view to understanding the structural correlates of neutralization of endotoxicity. We found clear differences in the inhibition of the LPS-induced cytokine production (Fig. 1) for the four lipopolyamines that we elected to characterize. DS96 has previously shown to be an excellent LPS neutralizer [12-14,29,30]. We were desirous of examining the relative contributions of the five cationic amine groups in DS96 (Scheme 1), and we therefore synthesized DS176 (Scheme 3), in which two of the internal secondary amines are replaced by amides, obviating cationic charges at these positions. We found that DS176 was significantly less active than DS96 (Fig. 1), emphasizing the importance of the potential salt-bridges between the internal secondary amines of the lipopolyamine and the Kdo groups in LPS as we had previously postulated [13,14]. Exhaustive Michael addition of DS96 yielded the analogue Yan673, in which all the amines are peralkylated (Scheme 1); this compound was found to be virtually inactive, pointing to the indispensability of protonatable amine groups for ionic H-bond formation in the complex. Replacement of the long-chain N-alkyl group with a biphenyl appendage in DS347 results in attenuation of neutralizing activity, indicating that an aliphatic alkyl group is optimal for hydrophobic interactions with the polyacyl domain of LPS [13,14].
The differences observed in the phase transition experiments (Figs. 2, 3), in which DS176 leads to a stronger fluidization of the acyl chains than compound DS96, appear not to correspond to neutralizing activity, since the active LPA DS347 has also high fluidization efficiency (Fig. 2, 3). This is an accordance to former data for the inactivation efficiency for different antimicrobial peptides, for example polymyxin B (PMB) and its nonapeptide PMBN [23], and newly developed compounds called synthetic anti-LPS peptides (SALP) [16]. The degree of fluidization of LPS superstructures is therefore not a main determinant, but could still be contributory to LPS inhibition. This is in contrast to what is observed for the aggregate structures of LPS, which may be organized as bilayered arrangements within cubic or hexagonal symmetry depending on the lengths of the sugar chains [27,28], but which never adopt a multilamellar organization except in cases of underacylated (tetra- or pentaacyl) lipid A moiety, which by themselves do not stimulate immune cells but rather may be able to antagonize the activity of active LPS [31,32]. The data presented in Fig. (4A) clearly indicate that the inactive DS176 does not cause any change of the LPS aggregate structure, while the addition of DS347 leads to a multilamellarization of LPS (Fig. 4B). This supramolecular arrangement of LPS was not only found for LPS and lipid A, which are inactive a priori, but was adopted in particular for all bioactive LPS and lipid A, which were inactivated by the addition of antimicrobial peptides or proteins [33-37]. A plausible explanation for these observations is that in large multilamellar aggregates, the regions within LPS responsible for the recognition by lipopolysaccharide-binding protein (LBP), CD14, and the TLR4/MD2 complex [38,39], are cryptic, obviating interactions of LPS with the cells and, consequently, leading to no cellular activation.
The thermodynamic outcomes of binding leading to the inactivation of LPS (Fig. 6) is particularly instructive. For both highly inactivating LPAs, there is a strong, saturable exothermic reaction with LPS, which runs into saturation within a sigmoidal curve at already low concentrations of the LPAs. Our interpretation of this behavior is that there is an initial polar interaction of the amine-containing aliphatic part with the backbone of LPS, followed by hydrophobic interactions of the hydrophobic moieties of DS96 (hexadecyl alkyl chain) or DS347 (biphenyl group) react with the polyacyl domain of lipid A. Such hetero-amphiphilic interactions have been shown to be very crucial for the interaction of LPS with the SALP [9,16]. Indeed, for the LPS-binding peptides, the first step consists of a direct Coulombic interaction of the positively charged amino acids of the SALP with the negatively charged LPS backbone groups (phosphates, carboxylates). This does not take place only in isolated LPS aggregates, but also with LPS or Gram-positive lipoprotein within outer membranes of the respective bacteria [40].
It has been shown that one of the properties of the SALP and other peptides consists of their ability to intercalate not only into membrane phospholipids such as PC and PS, but also into LPS aggregates [16]. Similar observations were noted with the lipopolyamines (Figs. 5), wherein the LPAs are incorporated into target cell membranes irrespective of their activity. In particular, the identical incorporation patterns of DS176 and DS347 into LPS aggregates (Fig. 5, top) suggest that the conversion of LPS from lamellar to aggregate superstructures (Fig. 4) correspond to biological activity of the LPAs.
CONCLUSIONS
We have brought to bear a variety of biophysical techniques to probe the interactions of LPS with candidate lipopolyamines which differ in their potencies of LPS neutralization. The results indicate that the ability to induce changes in supramolecular aggregate structure of LPS as a consequence of its interaction with the lipopolyamines is a biophysical correlate of endotoxin neutralization, and will be of value in the design and development of endotoxin sequestering agents from first principles.
ABBREVIATIONS
| LPS | =lipopolysaccharide |
| LP | =lipoproteins |
| LPA | =lipopolyamines |
| SALP | =synthetic anti-LPS peptides |
ACKNOWLEDGEMENTS
The authors are indebted to the National Institutes of Health (5U01 AI077947), the Bundesministerium für Bildung und Forschung (Project 01GU0824) and the Else-Kröner-Fresenius Stiftung (project 2011 A_140) for financial support.
REFERENCES
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]

