All published articles of this journal are available on ScienceDirect.

Parkin, A Top Level Manager in the Cell’s Sanitation Department
Abstract
Parkin belongs to a class of multiple RING domain proteins designated as RBR (RING, in between RING, RING) proteins. In this review we examine what is known regarding the structure/function relationship of the Parkin protein. Parkin contains three RING domains plus a ubiquitin-like domain and an in-between-RING (IBR) domain. RING domains are rich in cysteine amino acids that act as ligands to bind zinc ions. RING domains may interact with DNA or with other proteins and perform a wide range of functions. Some function as E3 ubiquitin ligases, participating in attachment of ubiquitin chains to signal proteasome degradation; however, ubiquitin may be attached for purposes other than proteasome degradation.
It was determined that the C-terminal most RING, RING2, is essential for Parkin to function as an E3 ubiquitin ligase and a number of substrates have been identified. However, Parkin also participates in a number of other fiunctions, such as DNA repair, microtubule stabilization, and formation of aggresomes. Some functions, such as participation in a multi-protein complex implicated in NMDA activity at the post synaptic density, do not require ubiquitination of substrate molecules. Recent observations of RING proteins suggest their function may be regulated by zinc ion binding. We have modeled the three RING domains of Parkin and have identified a new set of RING2 ligands. This set allows for binding of two rather than just one zinc ion, opening the possibility that the number of zinc ions bound acts as a molecular switch to modulate Parkin function.
I. INTRODUCTION
Genetic mutations in six genes have been implicated in hereditary Parkinson’s disease (PD). Mutations in any one of three genes, α-synuclein, leucine-rich repeat kinase 2 (LRRK2), or ATP13A2, give rise to autosomal dominant PD, whereas autosomal recessive PD occurs from mutations in PINK1 [PTEN (phosphatase and tensin homologue deleted on chromosome 10)-induced putative kinase 1], DJ-1, or the Parkin gene (reviewed in [1-3]). First identified in Japan, Autosomal Recessive Juvenile Parkinson’s disease (ARJP) has an early onset and results from mutations in the Parkin gene [4, 5]. Symptoms of ARJP resemble those of idiopathic PD although Lewy bodies, the diagnostic hallmark of PD, are absent in ARJP. Most studies of the Parkin protein have focused on its role in Parkinson’s disease; however, studies have also shown that Parkin participates in the clearance of the amyloidogenic Alzheimer’s disease Aβ(1-42) peptide [6-8] and in the clearance of the Huntington’s disease polyglutamine proteins [9, 10]. Parkin binding and ubiquitination of polyglutamine proteins is enhanced by hsp70 interaction with Parkin [9]. Both Aβ(1-42) and polyglutamine proteins impair proteasomal function, a common characteristic feature of several adult neurodegenerative diseases, emphasizing the importance of Parkin activity in maintaining the cellular ubiquitin proteasome system.
The Parkin gene is located on chromosome 6 (6q25.2-q27), is greater than 500 kb and contains 12 exons [4]. The Parkin gene coding sequence is in the GenBank database (accession number AB009973) and contains the nucleic acid base pair and protein amino acid residue numbering of Kitada, et al. [4]. The 4.5 kb parkin transcript contains a 1395 bp open reading frame that is translated into a 465 amino acid protein with a molecular weight of 51,652 daltons. Parkin is abundantly expressed in numerous parts of the brain as well as in heart and skeletal muscle, testis, stomach, adrenal gland, thyroid, and spinal chord [4].
The main focus of this review is to examine what is known regarding the structure/function relationship of the Parkin protein. By incorporating recent new information along with predicted Parkin protein structures, we hope to advance our understanding of how Parkin might function to alleviate toxicity of misfolded/aggregated proteins in PD and also in Alzheimer’s and Huntington’s diseases.
II. PARKIN DOMAIN STRUCTURE
Parkin belongs to the E3 ubiquitin ligase subset of the RBR protein family (reviewed in Eisenhaber et al. (2007) [11]. The primary domain structure of RBR ubiquitin ligases consists of three consecutive domains, each of approximately 50 amino acids: RING1, in between RING (IBR), and RING2. The Parkin RBR domain structure is shown in Fig. (1A). Historically, “RING”, an abbreviation for “Really Interesting New Gene”, was coined by Lovering et al. [12] and described a cysteine-rich motif. The combination of two linked RINGs in the same protein was first described by Van der Reijden (1999) [13] and eventually became known as the RBR. The RBR structure is apparently an ancient evolutionary feature that has endured for over a billion years [14]. Parkin orthologs, genes similar to Parkin because they originate from a common ancestor, can be found in vertebrates, insects and nematodes, but not in fungi or plants [14]. The amino acid residues, cysteine and histidine are interspersed among the RING amino acid sequences in a pattern that promotes binding of zinc ions. A sequence alignment comparison of RBR proteins revealed that the two RING domains in the RBR differed in spacing and number of conserved amino acid residues [14] (Fig. 1B). In general, RING1 is longer with more variability in spacing than RING2. The Parkin RINGs can be fit into this general sequence structure (Fig. 1A and B). RING1 holds two zinc ions in a cross brace arrangement with the first and second Cys pairing with the fourth and fifth Cys to hold one zinc ion (Fig. 1C). A second zinc ion is bound by the third Cys, the His, and paired with the sixth and seventh Cys, (reviewed in Borden (2000) [15] and Matthews and Sunde (2002) [16]).
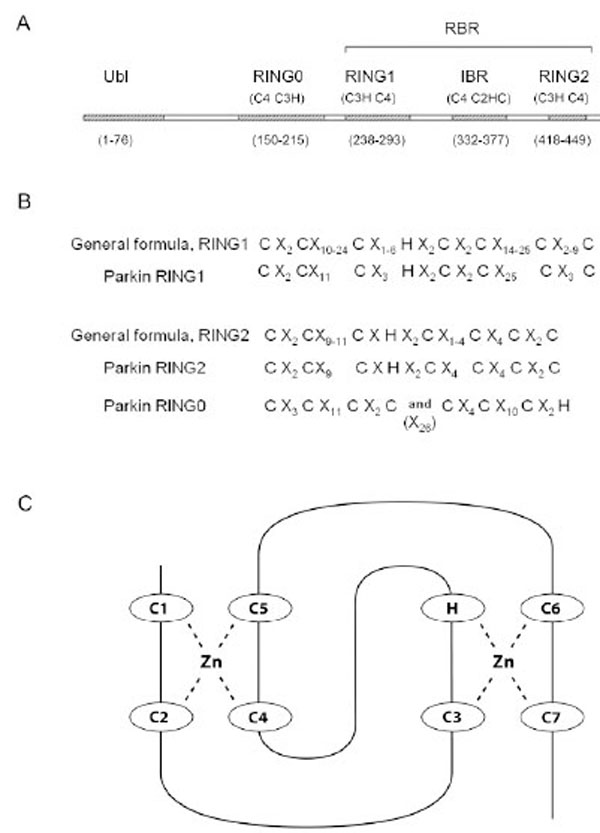
A) Domain Structure of the Parkin Protein. The five major domains of Parkin are listed across the top and include the Ubiquitin-like domain (Ubl), RING0, as well as RING1, the In Between RING domain (IBR), and RING2 within the Ring IBR RING, (RBR) region. Listed under each zinc binding domain is the pattern of each zinc ion binding site, i.e. RING0 site I = 4 Cys residues (C4), RING0 site II = three Cys and one His residue (C3H), etc. And, finally, the number of amino acid residues in each domain is listed at the bottom. Note that the RING2 pattern of Cys and His zinc binding ligands, as well as the domain boundaries listed at the bottom are according to the Morett and Bork (1999) [104] selection of ligands and not those predicted by our new RING2 model. B) The spacing of Cys and His zinc ion ligands in RBR proteins according to an alignment comparison by Marin and Ferrus (2002) [14]. X indicates the number of amino acid residues between consecutive ligands in the primary protein sequence. C) The classical cross-brace structure shows the interleaved ligand arrangement for the two zinc binding sites and is found in many RING domains. RING1 of Parkin likely forms a cross brace structure with parkin C238=C1, C241=C2, C253=C3, H257, C260=C4, C263=C5, C289=C6, and C293=C7.
The arrangement of conserved Cys and His amino acid residues in the IBR (the domain separating RING1 from RING2, see Fig. 1A) is split between two zinc-binding domains, one site containing four Cys (in Parkin, C332, C337, C352 and C360) and the other containing three Cys and one His (C365, C368, H373 and C377) [17]. The IBR domain structure of Parkin was determined using NMR spectroscopy and the zinc binding arrangement brings the N- and C-terminus of the IBR domain together with the result that RING1 and RING2 are brought into close proximity [17].
The structural determination of RING2 was problematic, leaving several unresolved issues. For example, a RING2 domain structural study of the closely related HHARI protein showed that only one zinc ion was bound and by analogy this was projected to Parkin RING2 [18]. However, in a Parkin zinc depletion experiment where zinc ions were sequentially removed by addition of dilute formic acid to denature Parkin, it was found that the total number of zinc ions bound by Parkin required that RING2 bind two zinc ions [19]. This, as well as other inconsistencies will be addressed in “Part VIII. Predicted RING structures”.
Recently a third RING domain was identified in Parkin and, as it is located upstream of RING1, was named RING0 [19] (Fig. 1A). The RING0 structural domain was identified by classic proteolytic biochemistry which is based on the premise that a stable, folded conformation limits proteolysis. A sequence comparison of human Parkin with Parkin of 12 other species showed a high degree of conservation through the RING0 domain and indicated the conserved Cys residues that are most likely involved in zinc binding [19]. RING0 apparently has two parts (C4 and C3H) separated by a 26 amino acid linker region (Fig. 1A and B). HOIL-1 is the only other RBR protein to contain a zinc binding motif upstream of RING1, however, it appears to bind only one zinc ion (discussed in Hristova et al. (2009) [19]. RING0 will also be discussed in more detail in “Part VIII: Predicted RING structures”.
Parkin contains 35 cysteines and binds a total of eight zinc ions, apparently two each in RING0, RING1, the IBR, and RING2 [19]. There are considered to be three reasons for proteins to bind zinc, structural, catalytic, and cocatalytic (reviewed in Auld (2001)) [20]. There is evidence that zinc binding directly affects Parkin structure both in regards to stability and for proper folding. Structural zinc sites contain 4 amino acids whose R groups can donate a pair of electrons for Zn++ binding. The amino acid Cys is the preferred electron donor with His as the second most prevalent donor.
In light of this, it is not surprising that the IBR domain of Parkin is completely unfolded by removal of zinc ions with EDTA [17], as is RING2 of the HHARI protein [18]. As zinc ions were removed by equivalents of EDTA the CD spectra of full length Parkin showed a gradual loss of secondary structure, i.e., α-helical or β-sheet [19]. However, it should be noted that removal of zinc by EDTA does not completely denature the protein; therefore, not all Parkin structure is dependent on binding of zinc ions [19]. Structural zinc binding can exert indirect effects on catalytic function of the protein by changing the chemical environment of the active catalytic site or by realigning important amino acids in that site [20]. In addition, certain Parkin mutations that disrupt zinc binding sites may also destroy E3 ubiquitin ligase activity by affecting the stability or solubility of Parkin [21].
One other Parkin domain, the ubiqitin-like domain (Ubl) located at the N-terminus, does not appear to be involved in zinc binding. The crystal structure of the Parkin Ubl shows a typical ubiquitin fold, containing 5 β-sheets and two α-helices, ββαββαβ [22]. From analysis of ARJP mutations in this domain it has been determined that the domain is stabilized both by hydrogen bonds and hydrophobic interactions [22]. One mutation, R42P, causes a change in the position of the β3 strand and completely unfolds the Ubl domain [22, 23]. Of interest is the discovery of an N-terminally truncated 42 kDa Parkin protein that is formed from an internal methionine start site (amino acid 80) [24]. This N-terminally truncated Parkin which lacks the Ubl domain was stable [24] suggesting that lack of the Ubl domain does not necessarily induce degradation of Parkin.
III. PARKIN MUTATIONS
Numerous naturally occurring Parkin mutations have been discovered and many of these are pathological, resulting in ARJP [4, 5]. The number of mutations, the location of the mutation and whether the mutation was heterozygous, all influence clinical manifestations [25]. An excellent ARJP mutational analysis was performed by Sriram et al. in which the characteristics of 12 ARJP mutations were determined in regards to substrate binding and E3 ligase activity [26]. Mutations have also been engineered so as to better understand how Parkin structure might affect Parkin function [27, 28].
C-Terminal Mutations Affect Parkin Stability
Parkin is aggregation-prone and is dependent on chaperones for proper folding [29]. A series of engineered deletion mutations involving the 16 C-terminal amino acids showed that truncations exceeding 3 amino acids produced insoluble Parkin [29]. An ARJP mutation in which tryptophan 453 is replaced with a termination codon (W453X), lacks the last 13 amino acids and is insoluble; however, the chaperone, hsp70, which prevents aggregation of wild type Parkin also promotes folding of the W453X protein [29]. Using proteinase K digestion, it was found that the conformation assumed by W453X is different from that of wild type Parkin [29].
The insolubility of C-terminal deletions is specific to Parkin and is not shared by the very similar HHARI [30]. Nor do the C-terminal amino acids from HHARI rescue a hybrid Parkin-HHARI construct as the chimeric protein was rapidly degraded by the proteasome [30]. In Parkin, the amino acid F463 was found to be highly conserved between species, and when mutated to Ala, was found to form insoluble aggregates [30]. The role of the C-terminus in Parkin stability will be addressed in further detail in “Part VIII. Predicted RING structures”.
N-Terminal Mutations
The mutation R42P is located in the ubiquitin-like domain and is often mentioned in the literature. It appears that R42P protein is highly unstable, being rapidly degraded by the proteasome. Only under conditions of proteasome inhibition, can R42P be detected and then it is found in the insoluble fraction. This suggests that R42P protein assumes a non-native conformation which is rapidly degraded [30]. Interestingly, an N-terminally truncated variant of Parkin found in human brain and cultured cells that resulted from translation initiation at an internal methionine residue at position 80 [24] was soluble whereas full length Parkin carrying the R42P mutation was not [27].
Mutation of Parkin Cysteine Residues
Since 28 of the 35 cysteines in Parkin are involved in zinc binding, it was of interest to determine what would happen if any of those cysteines were mutated. Mutational analyses revealed that the great majority of the Parkin Cys residues are essential to maintaining Parkin solubility [21, 27]. Cysteine mutations that did not appear to affect solubility were C59A, C95A, C268A, C323A, C431A and C451A [21].
Finally, there are two interesting experimental outcomes regarding mutations. In one experiment, a double mutation (R42P and W453X) was expressed in the same protein. The R42P alone resulted in proteasomal degradation of Parkin, i.e., instability, whereas the W453X mutation alone resulted in insoluble aggregates. On expression in SH-SY5Y cells, the double mutant Parkin was only detected with proteasome inhibition, and then only in insoluble aggregates. Therefore, proteasomal degradation was dominant over aggregation, i.e., no aggregates formed with an intact ubiquitin proteasome degradation system [30]. The second interesting experimental outcome revealed that some innocuously appearing mutations, i.e., ones showing no signs of decreased Parkin solubility, sensitized Parkin to cell-induced stress [31]. That stress might include oxidative stress, proteolytic (unfolded protein) stress, s-nitrosylation, and/or neurotoxins. Wang et al. (2005) [31] then summarized the types of mutations according to their effects on Parkin, the first type by directly reducing or destroying catalytic function, the second type by changing solubility and location, and the third type by increasing sensitivity to environmental stresses.
IV. PARKIN FUNCTIONS IN CELLULAR PROTEIN REMOVAL
Parkin functions as a top level manager in the cell’s Sanitation Department. The ubiquitin proteasome system (UPS) is the main cellular protein removal system (reviewed in Finley and Chau (1991) [32]); however, should the UPS become overwhelmed or in some way incapacitated, proteins to be removed may be sequestered in aggresomes for eventual autophagic clearance (reviewed in Johnston et al. (1998), Kopito (2000) [33, 34]).
Parkin and the Ubiquitin Proteasome System
In the UPS a small protein tag, ubiquitin, is covalently attached to the target protein through a series of steps, the last catalyzed by an E3 ubiquitin ligase. The first step is the ATP-dependent activation of ubiquitin by an E1 ubiquitin-activating enzyme. The activated ubiquitin is then transferred to a ubiquitin-conjugating enzyme, E2. The E3 ubiquitin ligase binds both the E2-ubiquitin and a specific protein target to which the ubiquitin is then covalently attached. Each step in the process becomes more restricted in its substrate, with the E3 ligase being the most specific (reviewed in Finley and Chau (1991) [32]; more recently in Deshaies and Joazeiro (2009) [35]). Parkin has been shown to have E3 ubiquitin ligase activity [36-39]. Consistent with a role in the ubiquitin proteasome system, Parkin has been shown to bind two subunits in the 26S proteasome, Rpn10 of the 19S regulatory subcomplex and the α4 subunit of the 20S proteolytic core [40, 41].
Ubiquitin is attached by formation of an isopeptide bond between the ubiquitin C-terminal Gly residue and the ε-amino group of a Lys residue in the targeted protein. There are at least three ways for a protein to be ubiquitinated: i) a chain of ubiquitin molecules formed by linkage to Lys 48 of the previously attached ubiquitin, ii) a chain of ubiquitin molecules linked through ubiquitin Lys 63, and iii) multimonoubiquitination, defined as multiple ubiquitin molecules per protein but attachment is one ubiquitin per lysine residue. Ubiquitin chains formed by Lys 48 linkage targets proteins for proteasomal degradation and a chain of at least four subunits is required for recognition and degradation [42]. Parkin can auto-ubiquitinate and in cell-free in vitro assays has been shown to multimonoubiquitinate itself [27, 43] as well as proteins that are in close proximity [43]. However, as discussed in the Hampe et al. report [27], the cell-free in vitro ubiquitination assays did not include other proteins that might be present in the cell. It was hypothesized that Parkin alone produces multimonoubiquitinated substrates whereas Parkin associated with other proteins conjugates polyubiquitin chains [27]. One of these other proteins may be an E4, (identified and described by Koegl et al. (1999) [44]. E4 ubiquitin ligases can be described as ubiquitin chain elongation factors. Without E4, substrate ubiquitination consists of only a few ubiquitin molecules, insufficient to accomplish proteasomal degradation [44]. Proteins such as Parkin, with ubiquitin-like domains at their N-terminus, are analogous to substrates of E4, (in particular of UFD2, a yeast member of the E4 family), and are likely to be degraded by the proteasome system [44].
Parkin has been shown to be auto-ubiquitinated when over-expressed in cultured 293, 293T and COS1 cells [36]; and co-transfection with the K48R ubiquitin mutation that prevents formation of K48- linked chains, inhibited Parkin ubiquitination [36]. This suggested that the ubiquitins were attached as Lys 48-linked chains. An unidentified mammalian E4 may facilitate K48-linked chains in cultured cells, but with no E4 in cell-free assays, Parkin is multimonoubiquitinated. Another study utilizing SH-SY5Y cells found that ubiquitinated Parkin could be identified only when cells were treated with the proteasome inhibitor MG132, suggesting that in neuronal cells with uninhibited proteasomes, ubiquitinated Parkin was rapidly degraded via the UPS [37].
As mentioned above, should the ubiquitin proteasome system become compromised in some way, proteins to be removed may be sequestered in the aggresome for eventual removal by macroautophagy. An aggresome is defined as a microtubule-dependent inclusion body that forms around the microtubule organizing center and is near to, or encompasses the centrosomes [33]. Proteins to be degraded by the aggresome pathway are actively transported along microtubules to the centrosome. At the same time, components of the UPS, e.g. ubiquitin, ubiquitin conjugating enzymes, proteasomal constituents, certain chaperones, and mitochondria are also recruited. These components are then organized into an aggresome (reviewed in Olanow (2004) [45]). Abnormal or damaged proteins are held within the aggresome by a cage of intermediate filaments, e.g. vimentin [33]. Parkin is actively involved in both the UPS and the aggresome system.
Parkin Substrates Ubiquitinated for Degradation
Of the many responsibilities assigned to the cellular Sanitation Department, Parkin participates either directly or by “committee” (as a member of a multi-protein complex) in removing i) short-lived proteins, ii) altered proteins, iii) excessive levels of proteins and iv) unfolded proteins. A short-lived Parkin substrate, SIM2, is a transcription factor located in the nucleus, and, like many short-lived proteins, contains a PEST degradation signal rich in the amino acids proline (P), glutamic acid (E), serine (S) and threonine (T). The human SIM2 protein is ubiquitinated by both Parkin and HHARI [46]. SIM2 is located in the nucleus whereas Parkin is found mainly in the cytoplasm [46]; however, the authors note that a very small amount of Parkin can be found in the nucleus and this nuclear Parkin is suspected of ubiquitinating SIM2 [46].
αSp22, a glycosylated form of α-synuclein, is an altered protein that is ubiquitinated by Parkin [47]. αSp22 accumulates in ARJP brain, but is undetectable in extracts from normal brain [47]. αSp22 appears to be unique to human brain as it could not be detected in lysates from HEK293 or SH-SY5Y cells transiently transfected with Parkin and α-synuclein, even when those cells were oxidatively stressed [48].
The toxicity of a third class of Parkin substrates, proteins that exceed certain concentration levels, was demonstrated by exogenous overexpression of these Parkin substrates in cell culture. Pael-R, p38, and Synaptotagmin XI are in this class of Parkin substrates. Pael-R is a G protein-coupled receptor thought to play a role in dopaminergic signaling. Over-expression of Pael-R produced insoluble, ubiquitinated protein and increased cell death [49]. Normal expression levels of Pael-R do not lead to insoluble accumulations in brain; however, insoluble Pael-R accumulates in ARJP brain [49]. Recently, a rat model of PD was developed by capitalizing on the fact that dopaminergic nerurons are especially sensitive to Pael-R accumulation [50].
The protein p38 is a structural component of the aminoacyl-tRNA synthetase complex involved in protein biosynthesis [51]. Over-expression of p38 in the dopaminergic cell line SH-SY5Y increased cell death by approximately 6-fold whereas over-expression of both p38 and Parkin reduced the amount of cell death when compared to p38 alone. This suggests that the toxic affect of increased p38 expression is partly alleviated by Parkin [51]. We assigned Synaptotagmin XI (SytXI), involved in the maintenance of synaptic function, to this category since a scan for SytXI mutations that might potentially produce disease in either inherited or sporadic PD was negative [52]. This suggested that protein levels rather than protein abnormalities were the key trigger for ubiquitination and degradation. However, this does not preclude SytXI degradation as a result of misfolding. In sporadic PD, SytXI is found in the core of Lewy bodies, suggesting that SytXI may be misfolded and targeted for degradation by the aggresome rather than by the proteasome pathway (discussed below). SytXI was identified as a Parkin substrate by yeast two hybrid followed by co-immunoprecipitation assays [53]. It was determined from the appearance of the bands on immunoblots that SytXI is polyubiquitinated and pulse-chase experiments using transiently transfected HEK293 cells showed that Parkin was responsible for the rapid turnover of SytXI.
Improperly folded proteins consititute a large number of proteins tagged for degradation. Upwards of 30% of newly synthesized proteins are improperly folded and the remaining 70% may at some later time be damaged or denatured during metabolic processes or by a change in the microenvironment of the cell (reviewed in Tanaka et al. (2004) [54]). Secretory proteins are folded within the ER; however, when the folding process fails, the unfolded proteins are removed to the cytosol for proteasomal degradation. Unfolded proteins produce “unfolded protein stress” which initiates a cellular “unfolded protein response”. One of the first steps in the unfolded protein response is an up-regulation of genes/proteins which aid in refolding or in protein degradation. One of these up-regulated genes, BiP, has become a marker for the unfolded protein response and was identified in the insoluble fractions of ARJP brain [49]. This suggests that the unfolded protein degradation pathway is activated in the absence of functional Parkin. An overwhelmed or impaired proteasomal system can also produce unfolded protein stress and the resulting unfolded protein response.
Parkin Activity in Response to Unfolded Protein Stress is Cell-Specific
Imai et al. (2000) [36] reported that Parkin specifically blocked cell death from unfolded protein stress and that Parkin activity was cell specific, i.e., the SH-SY5Y dopaminergic cell line had higher levels of protein ubiquitination than the HEK293 human embryonic kidney cell line. Parkin expression was up-regulated and cell death resulting from unfolded protein stress was reduced [36]. Although West et al. (2003) [55] were unable to confirm these results, a study utilizing primary cultures of rat hippocampal neurons and astrocytes showed that Parkin expression increased in the astrocyte, but not in the hippocampal cultures in response to unfolded protein stress [56]. Further, treatment of astrocytes with unfolded protein stress inducers resulted in formation of structures similar to aggresomes in appearance and location [56].
The “Lewy Body is a Failed Aggresome” Hypothesis
Lewy bodies are cytoplasmic inclusions that are found in sporadic PD (described in Olanow et al. (2004)) [45]. They are very similar to aggresomes, which form in response to unfolded protein stress or to an inefficient or inhibited proteasomal system (reviewed in Kopito (2000) [34]). Clearance of aggresomes may involve the macroautophagy system whereby the aggresome is engulfed by a double membrane to form an autophagasome which fuses with an endosome (or lysosome) to complete proteolytic digestion and clear the aggresome from the cell [34]. If aggresomal protein is not degraded, the structure may become an insoluble cellular inclusion. Due to the structural similarities between aggresomes and Lewy bodies, Olanow et al. 2004 hypothesized that Lewy bodies are failed aggresomes, i.e. they are unable to complete proteolysis successfully.
Recently, Wong et al. (2008) [57], presented evidence suggesting that aggresome clearance by macroautophagy is determined by the components recruited to the aggresome along with the aggregated protein. Some proteins, such as the Parkin substrate p38 fail to recruit key macroautophagy components to the aggresome and are therefore autophagy resistant [57].
Parkin Contributes to Lewy Body Formation
Synphilin-1 is an α-synuclein interacting protein found in Lewy bodies [48] and confirmation of Synphilin-1 as a binding partner and substrate of Parkin led to the finding that E3 ligase activity of Parkin contributes to Lewy body formation. Lewy bodies are by definition, ubiquitinated cytoplasmic inclusions. Co-expression of Parkin, synphilin-1 and α-synuclein resulted in ubiquitinated Lewy-body-like inclusions [48]. However, without expression of Parkin, these inclusions were not ubiquitinated and therefore by definition were not Lewy bodies. That Parkin contributes to Lewy body formation was further supported by the finding that Parkin localized to Lewy bodies [58] and that 90% of nigral Lewy bodies are immunopositive for Parkin [59]. Parkin mutations that result in loss of Parkin ubiquitination activity or that result in Parkin misfolding and subsequent degradation would obviously prevent Lewy body formation, thereby providing an explanation for the absence of Lewy bodies in ARJP.
Proteasome Inhibition and Aggresome Formation
It is important to keep in mind that most of the Parkin E3 ligase activity assays were performed in cultured cells and therefore much of the ubiquitinated substrate would be rapidly cleared by the UPS before samples could be collected. Detection could occur only if the amount of substrate completely overwhelmed the cellular UPS or if the UPS were inhibited. At this point it is unclear whether formation of aggresomes occured in response to an experimentally inhibited UPS or to a “normal” clearing process for that particular substrate. Two factors may help to clarify the issue: i) is the substrate found in Lewy bodies in idiopathic PD?, and ii) is the protein substrate membrane-associated?. Transmembrane or membrane-associated proteins are more likely to be misfolded and removed via the aggresome degradation system [33]. Parkin substrates synphilin-1, p38, and synaptotagmin XI are found in Lewy bodies whereas CDCrel-1 and Pael-R are examples of membrane-associated proteins.
Membrane-Associated Parkin Substrates
CDCrel-1 is a member of the human septin protein family and is recruited to the centrosome along with Parkin [60]. Although ubiquitin modified CDCrel-1 was detected in the absence of transfected parkin, levels increased with expressed Parkin and CDCrel-1 was found to be degraded in pulse-chase assays [38]. Originally thought to be associated with synaptic vesicles, more recent evidence suggests that only a small fraction of CDCrel-1 is found in synaptic vesicles [61]. Although the role of CDCrel-1 in Parkinson’s disease has not yet been clarified, a number of possibilities that lead to inhibition of dopamine release have been proposed [38]. Observations of CDCrel-1 null mice revealed no significant disruption of brain function, suggesting that other septins likely compensated for loss of CDCrel-1 [62]. Another septin, SEPT5_v2a is a close homolog of CDCrel-1 [63] and accumulated in 3 of the 4 ARJP brains examined [63].
Pael-R, a toxic protein when over-expressed (discussed above), was also reported to accumulate in the endoplasmic reticulum during unfolded protein stress. The up-regulation of BiP (marker gene for unfolded protein response), confirmed that the cells were responding to unfolded protein stress. Six hours after proteasome inhibition, Pael-R was found in the endoplasmic reticulum; after 16 hours, Pael-R was located within aggresome-like inclusions at juxtanuclear sites [49].
Although Parkin may be found in aggresomes/Lewy bodies, it may be sequestered for different purposes. Proteasomal inhibition resulted in aggresomal sequestration of wild type Parkin presumably to aide in the degradation of unfolded proteins [64], However, several Parkin mutations, e.g. R256C, R275W, C289G, and C418R which reduce Parkin solubility are relocated from cytoplasm to aggresomes, presumably to degrade the insoluble Parkin [21, 28, 65, 66].
Mechanics of Aggresome Formation and Microtubules
Proteins destined for aggresomal degradation are actively transported along microtubules to the centrosome as are components of the UPS, e.g. ubiquitin, ubiquitin conjugating enzymes, proteasomal constituents, certain chaperones, and mitochondria. These components are then organized into an aggresome (reviewed in Olanow et al. (2004)) [45]. When the cellular proteosomal system is inhibited, Parkin is recruited to the centrosome; removal of proteosomal inhibition reverses the centrosomal binding and Parkin is dispersed [60, 67]. This is accomplished by histone deacetylase 6 (HDAC6) which appears to sense proteosomal efficiency [67]. Generally, ubiquitinated proteins that are transported to the centrosome bind to the C-terminal ZnF-IBP motif of HDAC6 (discussed in Jiang et al. (2008)) [67]. Parkin however, binds to two other regions in HDAC6, DD1 and DD2. The interaction between Parkin and HDAC6 is direct and not mediated by microtubules. Recruitment of Parkin to the centrosome was shown to be dependent on HDAC6/dynein motor protein binding, and dispersal dependent on HDAC6 switching to the kinesin 1 protein (KIF5) [67]. HDAC6 deacetylates tubulin, and although apparently not acting directly on Parkin, the tubulin deacetylase activity of HDAC6 is required for Parkin recruitment to and dispersal from the centrosome [67].
Considering the importance of microtubules for centrosomal recruitment of Parkin and substrate proteins, it comes as no surprise that Parkin, as a top level manager in the cell’s Sanitation Department takes some responsibility for microtubule quality control and stabilization. Microtubules are formed from α/β tubulin heterodimers, and monomer levels of α and β tubulin are closely regulated so that neither is in excess. In addition, correct folding of tubulin is a complex process that results in a high level of misfolded tubulin (discussed in Ren et al. (2003)) [68]. Parkin co-immunoprecipitated with α or β-tubulin monomers in HEK293 cells and in rat brain lysates, and with α/β tubulin heterodimers in rat brain lysates [68]. A third type of tubulin, ɣ-tubulin, co-localized with Parkin in Lewy bodies (reviewed in Olanow et al. (2004) [45], McNaught et al. (2002)) [69].
Parkin interacts with microtubules, binding very tightly even in the presence of 3.8 M NaCl or 0.5 M urea and acting as a MAP (microtubule-associated protein [70]). Microtubules were stabilized when bound to wild type Parkin in the presence of the microtubule de-polymerizing agent, colchicine [70]. Parkin alone protected against the microtubule de-polymerizing PD toxin, rotenone, as shown by loss of protection in β-lymphocyte and fibroblast cell cultures derived from PD patients carrying Parkin mutations [71]. Parkin/microtubule interactions that function to stabilize microtubules do not appear to interfere with E3 ligase activity [68].
V. OTHER PARKIN-PROTEIN INTERACTIONS
Parkin interacts with a number of proteins that are not substrates for ubiquitination and are not targeted for degradation although they may be involved indirectly in protein quality control. These proteins may share properties with those in categories described above, or they may have some other important feature, i.e. location, function or, in the case of an enzyme, activity, that warrants a class of its own. A number of interacting proteins form multi-member protein complexes with Parkin. According to Marin and Ferrus (2002) there are six unrelated types of proteins or protein complexes with E3 ligase activity [14]. These proteins may have E3 ligase activity by themselves or as members of multi-protein complexes; the SCF, APC and VCB-like complexes are well characterized examples (reviewed in Tyers and Williams (1999)) [72]. In these complexes, RING structures play a central role both in complex formation and in E3 ubiquitination activity. In some cases the E3 ligase activity of Parkin may be self-contained, i.e. no proteins other than ubiquitin, E1 and E2 are required for E3 ligase activity [27, 39], in other cases Parkin may function as a part of a multi-protein complex. We will discuss these interacting proteins in this section.
BAG5, a component of Lewy bodies in sporadic PD interacts with hsp70 and with Parkin [73]. BAG5 inhibits the chaperone activity of hsp70 and, independent of hsp70, the E3 ligase activity of Parkin. Co-transfection of Parkin and hsp70 prior to proteosomal inhibition reduced the recruitment of Parkin to the centrosome, presumably because chaperone activity of hsp70 successfully refolds proteins and they do not require degradation. However, triple transfections of Parkin, hsp70, and BAG5 prior to proteasomal inhibition nullified the effect of hsp70, and Parkin was recruited to the centrosome [73]. BAG5 can therefore promote Parkin sequestration and enhance Lewy body formation. BAG5 also inhibits the decrease in cell death effected by Parkin when the ubiquitin proteosomal system is overwhelmed. BAG5 is expressed in dopaminergic neurons in the substantia nigra, and mRNA and protein levels are increased in response to neuronal injury. Expression of BAG5 promotes neurodegeneration in dopaminergic cells of the substantia nigra when expressed in rat brain [73].
Similar to ubiquitin, small ubiquitin-like modifiers known as SUMO can be attached to proteins as a signal. Also similar to ubiquitin, SUMO molecules are attached to their target protein by an E3 ligase specific for SUMO. RanBP2 is a member of a small ubiquitin-like modifier (SUMO) E3 ligase family and a substrate of Parkin. Carboxy-terminal glycine residues in the SUMO proteins are linked to lysines in the target protein via isopeptide bonds. A complex chain of events involving Parkin, Ran BP2, HDAC4, histones, chromatin condensation, and transcription allows Parkin to indirectly control transcription. RanBP2, is a 356 kDa component of the nuclear pore complex, in particular of the nuclear pore cytoplasmic filaments where disassembly of export complexes occurs [74], and RanBP2 binds to both Parkin and HDAC4 [75]. Levels of RanBP2 are controlled by Parkin ubiquitination and proteasome degradation. RanBP2 in turn regulates levels of HDAC4 by sumoylation [75]. HDAC4 is a Class II deacetylase and therefore shuttles between the cytoplasm and the nucleus through the nuclear pore complex (discussed in detail in Um et al. (2006)) [75]. HDACs deacetylate histones thereby promoting chromatin condensation and repressing transcription. Through control of RanBP2 levels it is possible for Parkin to indirectly control transcription.
It was also found that Parkin binds specifically to SUMO-1 (there are three classes of SUMOs, SUMO-1, -2, and -3), and although the binding is not a covalent modification it is sufficient to promote Parkin auto-ubiquitination and to transport Parkin from the cytoplasm to the nucleus [76]. Determining a mechanism for Parkin transport to the nucleus was an important finding considering the nuclear location of some Parkin substrates, e.g., SIM2 [46], Cyclin E [77], and poly-(Q)-expanded huntingtin [9].
Parkin associates with proliferating cell nuclear antigen, PCNA, a protein that coordinates DNA repair [78]. The parkin gene is located in a genomically unstable region (detailed in Kao (2009)) [78], and as a result, Parkin mutations, deletions and duplications are found in carcinomas of breast, lung, ovary and liver. Experiments utilizing SH-SY5Y cells stably transfected with control vector, wild type or mutated Parkin were irradiated [78] and expression of wild type Parkin was found to increase the number of surviving colonies. PCNA was implicated as a Parkin binding protein by co-immunoprecipitation [78]. PCNA interactions are mediated by a PCNA interacting peptide domain (PIP) and Parkin contains an imperfect, but functional, PIP domain in RING1 [78]. Parkin did not ubiquitinate PCNA in this study and several possible functions resulting from the Parkin/PCNA interaction were suggested, all of which would depend on how Parkin affected the binding properties of the PCNA complex [78]. It was found that DNA damage relocated Parkin to the nucleus [79].
Cyclin E is a regulatory subunit of cyclin-dependent kinase 2 protein that is highly expressed in adult brain neurons. Parkin forms a multi-protein complex with proteins, hSel-10 and Cullin-1, that, along with the E2, UbcH7, ubiquitinates Cyclin E. Increased levels of Cyclin E result in neuronal cell apoptosis in cells exposed to excitotoxic stress. hSel-10 contains the F-box and the WD repeat, common protein binding domains in E3 ubiquitin ligase complexes, although only the F-box domain is required to form the Parkin complex (detailed in Starpoli et al. (2003)) [77]. hSel-10 is an intermediate in the Parkin/Cullin-1 association since Parkin fails to bind Cullin-1 in the absence of hSel-10. An ARJP RING1 mutation, T240R, inhibits interaction of Parkin with hSel-10 and consequently Cullin-1. As a Parkin substrate, Cyclin E accumulates in the absence of Parkin. Regulation of Cyclin E by Parkin, plays a role in glutamate excitotoxicity as Parkin was shown to protect midbrain dopamine neuron cultures from kainate excitotoxity [77].
Interestingly, the three proteins involved in autosomal recessive PD pathogenesis, PINK1, DJ-1, and Parkin, form a three protein complex that functions to promote degradation of unfolded proteins via an E3 ligase activity that is more robust than that of Parkin alone [80]. ARJP may result from mutations in any one of these three genes. Parkin, PINK1, and DJ-1 interact via distinct domains and form a 200 kDa complex [80]. Perhaps the most significant aspect of this study is that participation in this complex appears to completely re-direct the function of PINK1 and DJ-1. Alone, both are involved with mitochondria. Although PINK1 alone is found in the mitochondria (and Parkin has been shown to alleviate aberrant mitochondrial morphology that occurs when PINK1 is absent or mutated [81]), it was shown that a 55kDa PINK1 fragment co-precipitated with Parkin. The complete PINK1 protein localized to the mitochondria whereas the 55kDa fragment that binds to Parkin was mainly in the cytosol [80].
During cell division, mitochondria are fragmented into hundreds of spherical vesicles to allow partitioning of mitochondrial fragments into the two daughter cells [82]. This process of mitochondrial division and fusion also happens during synapse formation in hippocampal neurons where mitochondria are recruited into neural protusions [83]. PINK1 is required for the normal transition between mitochondrial division and fusion [84]. From observation of DJ-1 knockout mice, the DJ-1 protein likely functions in the removal of hydrogen peroxide from mitochondria [85]. However, when combined into a PINK1, DJ-1, and Parkin complex, the assembly functions to degrade unfolded proteins [80].
The function of another multi-protein complex, CHIP, hsp70, and Parkin determines the fate of unfolded proteins. In this complex, Parkin appears to play the role of committee chair, recruiting committee members to make and carry out protein triage decisions. One committee member, hsp70, is a chaperone and functions in ATPase-dependent cycles of binding and release from small hydrophobic regions of misfolded proteins (reviewed in Bukau and Horwich (1998) [86]). Upon release, the protein may refold into its proper native structure, may bind hsp70 again, may aggregate, or may bind to other chaperones or preoteases. Imai et al. (2002) [87] report that hsp70 transiently binds to the complex substrate, in this case unfolded Pael-R, while, at the same time inhibiting Parkin E3 ligase activity. This prevents two conflicting processes, hsp70-directed refolding into proper native structure and ubiquitination prior to degradation, from happening simultaneously.
CHIP (carboxyl terminus of the Hsc-70 interacting protein) functions as a triage protein, directing proteins either for chaperone refolding or for proteasome degradation [88, 89]. Specifically, CHIP inhibits hsp70 substrate binding and refolding activities [90, 91]. In the CHIP, hsp70, Parkin, Pael-R complex, CHIP dissociates hsp70 from Pael-R, and thereby relieves Parkin E3 ligase inhibition so that Pael-R is ubiquitinated. This is accomplished through a higher binding affinity between Parkin and CHIP than between Parkin and hsp70 [87]. Consistent with the order of activity described above, i.e., binding of chaperone, followed by triage and ubiquitination, SH-SY5Y cells showed a transient increase in Pael-R-hsp70 association at 6 hours after induction of unfolded protein stress followed by a marked decrease in association at 24 hours [87].
Both CHIP and the chaperone hsp70 bind to wild type Parkin as well as any Parkin mutants containing an intact RING1 [87]. Interestingly, CHIP contains a U box (with similarity to yeast protein UFD2) that may, in vivo, act as an E4, elongating ubiquitin chains and directing unfolded proteins for degradation by the UPS (Koegl et al. (1999) [44] and discussed above). However, with proteasomal inhibition, e.g. in SH-SY5Y cells treated with the proteosome inhibitor lactacystin, CHIP, Pael-R and Parkin localized to aggresomal structures [87].
PDZ (postsynaptic density-95, disc large, zona occludens) domains are approximately 90 amino acids in length and interact with a specific short peptide sequence generally located at the C terminus of the ligand (reviewed in Sheng and Sala (2001)) [92]. The C-terminus of Parkin contains one of these specific short peptide sequences, (amino acids FDV) and has been found to interact with the PDZ protein, CASK [61]. CASK is a member of a large multi-protein complex that in rat brain, co-localized with Parkin within post synaptic densities and in the Triton X-100-insoluble lipid raft membrane compartment [61]. CASK is not a substrate for Parkin ubiquitination. More likely CASK serves as a link to formation of a large multiprotein complex, especially as Parkin co-immunoprecipitates, but does not bind directly to members of the complex which includes the PDZ protein PSD-95, the NMDAR subunit NR2B, the calcium and calmodulin-dependent protein kinase II (CaMKII), and homer 1a. These proteins are implicated in NMDA activity at the post synaptic density [61].
More recently it was discovered that a small amount of Parkin associates with membranes regardless of whether the PDZ peptide sequence is intact [30]. Further, disruption or removal of the PDZ peptide sequence does not impair Parkin’s neuroprotective function following kainate treatment, suggesting that protection from excitotoxicity does not reside solely in the Parkin/CASK interaction [30].
At least three E2 ligases bind to the Parkin RING2. These include UbcH8 [17, 38], UbcH7 [37] and the E2 ligase UbcH13 which dimerizes with Uev1a (UbcH13/Uev1a) and mediates Lys 63-linked ubiquitin chain formation thereby targeting proteins to the aggresome rather than UPS for degradation [93-95].
VI. REGULATION OF PARKIN
Parkin catalytic activity is negatively regulated by hsp70 in the CHIP, hsp70, Parkin complex as well as by BAG5 [73, 87]. And Parkin E3 ligase activity is positively regulated by the three protein complex, PINK1, DJ-1 and Parkin [80].
In another regulatory pathway, a chaperone-like protein, 14-3-3ɳ, binds to Parkin, negatively regulating both substrate binding and Parkin E3 ubiquitin ligase activity [96]. 14-3-3ɳ, binds to Parkin RING0 which contains the amino acid sequence RKDSPP (amino acid residues129-134), a typical binding motif for 14-3-3 proteins. α-synuclein and Parkin compete for 14-3-3ɳ, therefore an increase in α-synuclein concentration relieves the inhibition on Parkin-substrate binding and E3 ligase activity. The authors suggest that under conditions of low α-synuclein concentration, Parkin-14-3-3ɳ, binding allows large quantities of inactive Parkin to be retained in the cell by inhibiting the self-ubiquitination and proteasomal degradation that is observed in cultured cells over-expressing Parkin. As levels of α-synuclein rise and compete for 14-3-3ɳ, Parkin-substrate binding and ubiquitation activity would be restored [96].
Parkin activity is also regulated by reversible phosphorylation. Phosphorylated Parkin shows decreased E3 ligase activity suggesting that phophorylation down-regulates ubiquitination [97]. In the presence of ER unfolded protein stress, phosphorylation is reduced and E3 ligase activity increased. Proteasomal inhibition also reduces Parkin phosphorylation, although not as much as unfolded protein stress. Reduction of phosphorylation is specific to proteosomal dysfunction and ER stress; oxidative stress did not alter Parkin phosphorylation levels [97].
Parkin is constitutively phosphorylated although rapid turnover requires use of the phosphorylation inhibitor, okadaic acid, to identify phosphorylated Parkin [97]. Five serine phosphorylation sites, S101, S131, S136, S296. and S378 were identified and evidence of weakly phosphorylated threonine residues were also observed. These five serine residues are in the spacer between the Parkin Ubl and RING0 (S101, S131, S136); between RING1 and the IBR (S296); and between the IBR and RING2 (S378). Mutational analysis of the 5 serine sites confirmed that these were not the only sites phosphorylated in Parkin, and also that none of the 5 serine sites appeared to have a unique regulatory role [97]. Kinases likely involved in Parkin phosphorylation were identified as CK-1, PKA, and PKC.
S-nitrosylation can also regulate Parkin E3 ligase activity [98, 99]. Regulation is biphasic with activity increasing within a few hours of treatment with S-nitrocysteine and decreasing 12-24 hours later [99]. When SH-SY5Y cells over-expressing Parkin were incubated with S-nitrosocysteine, S-nitrosothiol formed on Parkin [99]. By use of peptide mass fingerprinting, (Q-TOF) MS, five of seven Cys residues in RING1 and potentially five Cys residues in RING2-IBR were shown to have undergone S-nitrosylation modification [99]. Mutational analyses of cysteines in the IBR showed that no single mutation eliminated S-nitrosylation, implying that multiple Cys residues are involved; quantitative analysis revealed that approximately three Cys residues per Parkin molecule are S-nitrosylated [98].
Interaction with SUMO-1 regulates both Parkin auto-ubiquitination and Parkin localization to the nucleus [76]. Since Parkin also interacts with RanBP2, an E3 SUMO ligase, Um and Chung questioned whether Parkin might be a substrate of RanBP2. However, SUMO-1 is not covalently attached, and therefore Parkin is not a substrate of RanBP2 [76].
With regard to structure vs function, regulatory activity appears to be spread over most of the Parkin molecule. Several regulatory factors appear to interact with RING0, e.g., PINK1 [80], 14-3-3ɳ [96] , and BAG5 [73]. Confirmed phosphorylation sites are spread from RING0 through the IBR, and nitrosylation occurs throughout the RBR region.
VII. PARKIN-PROTEIN INTERACTIONS AND E3 UBIQUITIN LIGASE ACTIVITY
Parkin-Protein Interactions
Generally, Parkin binding sites were determined by immunoprecipitation of the Parkin substrate or interacting protein using various fragments of Parkin. Several studies used different Parkin domains, parts of domains, or combinations of domains in their binding assays, thereby making comparisons between reported Parkin/protein interaction sites difficult. Keeping this in mind, certain generalizations regarding Parkin-protein binding can be made.
One important observation is that Parkin-protein interaction sites are spread over the entire Parkin molecule (Table 1). RING2 appears important for many proteins, either as the single binding site, as a part of a larger binding site, or as an alternative binding site. RING2 alone appears sufficient for binding Synphilin 1 [48], Pael-R [49], and UbcH13 [95] SIM2 [46] and α4 [41] require the adjacent IBR in addition to RING2, and the entire RBR is needed for DJ-1 [80] and hSel-10 [77]. Some proteins interact with RBR domains other than RING2. PCNA binds to a fragment containing both RING1 and the IBR (although the two domains were not tested separately) [78]; and both CHIP and hsp70 bind to RING1. Several proteins bind to domains outside of the RBR. RING0 is an alternative site for tubulin [70] and HDAC6 binding [67]; BAG5 appears to be the only protein that interacts solely with RING0 [73]. Rpn10 appears to be the only protein discovered thus far that interacts only with the ubiquitin-like domain [40]; Sept5_v2a interacts with either the ubiquitin-like domain or RING1-IBR of the RBR region [63].
Parkin-Protein Binding Sites. Sites in Parkin which have been Shown to Associate with other Proteins
| Parkin | Ubl (1-76) | RING0 (150-169) (196-215) | RING1 (238-293) | IBR (332-377) | RING2 (418-449) | PDZ | Referencs |
|---|---|---|---|---|---|---|---|
| synphilin 1 Pael-R UbcH13 | yes | [48] [49] [95] | |||||
| UbcH8 | ? | yes | [38] | ||||
| UbcH7 | yes | yes | ? | [37] | |||
| CDCrel-1 | yes | +yes | or yes | [38] | |||
| Tubulin HDAC6 | yes | yes | yes | [70] [67] | |||
| Cyclin E (hSel-10) | WD repeat (not required) | yes F-box | yes F-box | yes F-box | [77] | ||
| SIM2 | yes | +yes | [46] | ||||
| α4 | yes | +yes | [41] | ||||
| Synaptotagmin XI | yes (204-238) | yes (257-293) | [53] | ||||
| CHIP | yes (+ ?) | [87] | |||||
| hsp70 | yes (+ ?) | [87] | |||||
| PCNA | yes | +yes | [79] | ||||
| BAG5 | yes | [73] | |||||
| RanBP2 | yes (78- | 170) yes | [75] | ||||
| Rpn10 | yes | [40] | |||||
| PINK1 | (93- | 212) yes | [80] | ||||
| DJ-1 | yes (217-465) | +yes | +yes | [80] | |||
| CASK | yes | [61] | |||||
| p38 | yes (bait) | [51] | |||||
| Sept5_v2a | yes or | yes | +yes | [63] | |||
| 14-3-3η | yes | [96] |
The Parkin domains, Ubl, RING0, RING1, IBR RING2 and the specific short peptide sequence that binds PDZ domains are listed in the top row of the table. The proteins that associate with Parkin are listed in the left hand column. An empty box under a particular parkin domain means that domain has not been implicated in binding to the protein listed in the left column. A “yes” indicates that the protein listed on the left binds to that particular Parkin domain. A “?” indicates that the data is unclear in regard to whether the protein binds to that parkin domain. For instance, RING1, although not required for UbcH8 binding to Parkin, may enhance the binding interaction. A “+ yes” indicates that this domain is required in addition to another domain. A “+ ?” means that another unidentified domain is also needed . “Or yes” indicates an alternative binding site.
E3 Ubiquitin Ligase Activity
A number of studies have suggested that requirements for binding are less stringent than are requirements for ubiquitination or degradation of Parkin substrates. There are some general “rule of thumb” guidelines involving mutations that can be useful in examining binding sites. For instance, if a mutation is known to change the conformation of a molecule and also affects binding properties, then the binding site may be dependent on the conformation of the molecule. If the binding site requires non-consecutive regions of the molecule, then likely the site requires a structure that pulls those disparate regions together. And finally, if a binding site is a small specific primary sequence of amino acids, then the tertiary structure is less likely to affect binding unless the site is obscured by the tertiary structure. In such cases a mutation may actually increase binding if it denatures that small region of the molecule, perhaps by disruption of zinc ion binding that stabilizes the obscuring structure. Examples include: i) synphilin-1 which maps to RING2. As expected, familial-PD mutations in RING1 appeared to have no effect on binding; however, those in RING2 and the IBR actually increased binding [48]. At this point it is unclear why binding would increase. Although the ARJP mutations T240R and R256C (in RING1) as well as the truncating mutation W453X either had no effect or actually increased binding, ubiquitination of synphilin-1 was adversely affected by all three of these mutations [48]. Interestingly, a later investigation of the effect of mutations on Parkin E3 ligase activity identified the mutation T240R as belonging to a set of mutations (K161N, T240R, Q311X, T415N and P437L) that are considered “ligase dead” since they neither auto-ubiquitinate nor substrate ubiquitinate [26]. R256C belongs to a set (R256C, G328E, G430Q, C431F, and W453X) that auto-ubiquitinates but not substrate ubiquitinates [26]; ii) tubulin is bound, but not ubiquitinated, in the presence of Parkin mutations K161N in RING0, T240R in RING1, and C431A in RING2 [68]. Again, these belong to the set of “ligase dead” mutations [26]; iii) binding and ubiquitination of Parkin substrates (CDCrel-1, p38 and α-tubulin) or associated proteins (ɣ tubulin, hsp70 and the α4 subunit of the proteosome) were tested with ten Parkin mutations A82E, K161N, K211N, R256C, G328E, R42P, R275W, C289G, C418R, and C441R [27]. The binding interactions between these proteins and mutant Parkin variants did not appear to vary significantly from that of wild type Parkin. However, utilizing a cell-free in vitro ubiquitination assay that measured Parkin auto-ubiquitination, only C418R and C441R (which disrupt zinc ion binding in RING2) inhibited ubiquitination. Since the assay involved Parkin auto-ubiquitination we would expect mutations R256C and G328E (in the set of mutations that auto-ubiquitinate) to be unaffected; however, K161N was not “ligase dead” as might be expected.
It should be noted that there are many discrepancies in reports of substrate ubiquitination, some likely due to effects of other proteins in cell-based assays. And “in vitro” assays utilizing recombinant Parkin purified from reticulocytes may be affected by protein contaminants. This was discussed thoroughly in the Hampe et al. report [27].
General vs Specific Ubiquitination in the Presence of Parkin
Although Parkin expression increased ubiquitination of α- and β-tubulin in HEK293 cells, the authors also showed data suggesting that exogenous expression of Parkin may enhance ubiquitination levels for many different proteins [68]. Certainly, there is little doubt that Parkin recognizes specific proteins for ubiquitin modification; however, it is possible that Parkin alone or as a component of a multi-protein complex, perhaps with chaperone complex members, can recognize something more general such as exposed hydrophobic regions. Whether Parkin switches between a specific substrate to more general substrates, and if so, determining the mechanism for such a substrate switch, is an interesting question for possible exploration. Similarly, many questions remain regarding the circumstances under which a Parkin substrate is “chosen” for binding and/or ubiquitination,
VIII. PREDICTED RING STRUCTURES
Structure
The three RING domains (0, 1 and 2) of the Parkin protein were modeled using the I-TASSER program that generates structure predictions by first identifying templates (proteins with similar fold) for the query protein from the Protein Data Base (PDB) library. I-TASSER then assembles the fragments derived from the templates into full-length models using the parallel replica-exchange Monte Carlo simulations [100].
The models generated for each Parkin domain were analyzed to identify plausible locations of zinc ion binding sites based on sequence alignment and the position of zinc coordination sites in templates identified by threading algorithms. Zinc ions were added near the plausible binding sites and the structure was refined using the Gromacs molecular dynamics simulation package [101] for the purpose of refining the local structure of the backbone and side-chain atoms. The position of ligand residues in modeled structures of the three RING domains (0, 1, and 2) suggest that each domain can bind two zinc ions. We also modeled the Ubl and the IBR domain of Parkin. However, as our results are in agreement with the known structure of other ubiquitin conjugating proteins and with those presented earlier by Tomoo et al. (2008) [22] and Beasely et al. (2007) [17], we will not present our results here.
Model of RING1
The fold of RING1 closely matches the RING finger domain of human UbcM4-interacting protein (PDB: 1WIM). The predicted structure of the RING1 domain (Fig. 2A) and the threading alignment with the identified template, suggest that RING1 adopts the classical zinc ligand spacing [12, 102, 103] (Fig. 1B, 2B) and assumes a typical cross brace structure (Fig. 1C) with residues C238, C241, C260 and C263 as ligands in the first zinc co-ordination site and C253, H257, C289 and C293 present in the second site.
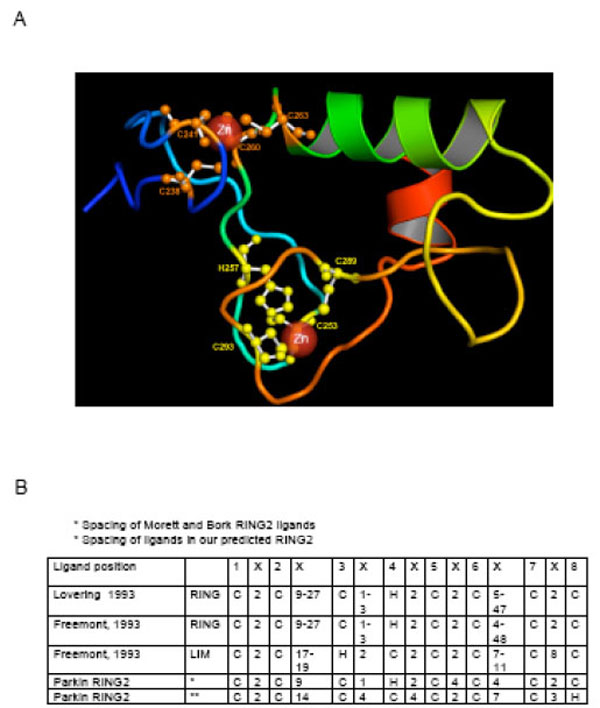
The predicted model of the RING1 domain using the I-TASSER algorithm. The model has a typical cross-brace structure and is in agreement with the previously suggested structure for Parkin RING1. The template identification was conducted using LOMETS, a metathreading server appoach with 9 third-party threading programs installed locally [111]. Multiple templates were collected from the topranking alignments in each of the 9 individual programs in LOMETS. A composite force field, consisting of the inherent knowledge-based potential and the spatial restraints collected from the multiple template structures, is then used for guiding the fragment assembly Monte Carlo simulations. The final models were selected based on the frequency of occurrence of the conformation appearing during the I-TASSER reassembly simulations (or the cluster size), identified by SPICKER [112]. Finally, full-atomic models are built by REMO [113] by optimizing the hydrogen-bonding network. B) The spacing of zinc site ligands in RING and LIM structures as described by Lovering et al. [12] and Freemont [103]. The ligands are numbered from one to eight in the top row and the intervening amino acid residues are depicted as “X”. The ligand spacing and location of the His residue is nearly the same in the two RING descriptions. The LIM structure is different in ligand spacing and also in location of the His residue.
Model of RING0
Our predicted structure for the RING0 domain is different from the previously proposed model. For example, a model for RING0 structure was proposed by Hristova. et al. consisting of two zinc binding sites containing (C4 and C3H) separated by a linker region (Fig. 1A). Because the linker region separating the zinc binding motifs is very large (26 amino acid) and because HOIL-1, the only other RBR protein to contain a zinc binding motif upstream of RING1, has only one zinc binding motif, Hristova et al. (2009) [19] proposed a model in which RING0 binds two zinc atoms in an independent and linear fashion rather than in a cross brace structure. However, a clear picture of the binding mode of zinc ions in the RING0 domain remained elusive.
Structure modeling of RING0 using the automated fold recognition procedure for RING0 did not identify any significant templates and therefore the model quality was low because the templates had diverse folds. Accordingly, the template proteins in the PDB library were filtered based on the presence of bound zinc ions and a linker region of at least 20 residues between the zinc coordination sites. The search resulted in identification of three DNAJ/hsp40 cysteine rich domains (PDB: 1exk), with sequence conservation mainly at/or near ligand binding sites.
The overall topology of the predicted model for RING0 (Fig. 3A and B) is based on the DNAJ/hsp40 homology and differs from that proposed by Hristova et al. (2009) [19] in that the two zinc binding sites are interleaved, with a V-shaped extended β-hairpin topology in the linker region, similar to the cysteine rich domain of DNAJ. The long linker apparently does not impair binding of zinc ligands. The rest of the domain contains only a limited amount of regular secondary structure. The first binding site consists of Ligands one (Cys 150), two (Cys 154), seven (C212) and eight (H215), while the second is composed of Ligands 3-6 (C166, C169, C196, and C201). While the predicted model requires experimental validation, it provides the first plausible three dimensional model for zinc binding in RING0.
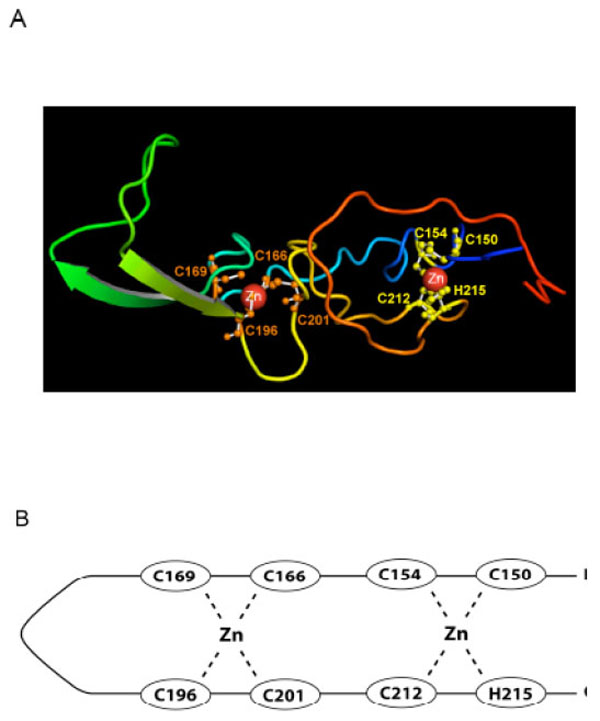
A) Our predicted structural model of the RING0 domain. The ligand residues shown in yellow (C150, C154, C212, and H215) represent the first zinc ion binding site; those in orange (C166, C169, C196, C201), the second binding site. B) A line drawing of our predicted structure shows the two interleaved zinc ion binding sites. Ligands one and two pair with seven and eight to form the first zinc binding site and the four internal Cys residues form the second zinc site. This is different than the RING0 structure predicted by Hristova et al. [19] where the first four Cys ligands form the first zinc site and the last three Cys plus the His residue form the second zinc binding site.
Model of RING2
A current model for RING2 structure is based on zinc ion ligand residues C418, C421, C431, H433, C436, C441, and C449 identified by Morett and Bork (1999) [104]. The model fits the classical ligand spacing [12, 102, 103, 105] (Fig. 2B), however spatial limitations prevent it from easily fitting into a cross-brace structure (Fig. 1C). In the Morett and Bork model, C418, C421, C431, H433 coordinate to the first zinc ion, but Capilli et al. (2004) showed that the RING2 domain of HHARI, a protein that shares both the RBR domain structure and the E3 ligase function of RING2 with Parkin, did not form a cross brace structure and binds only one zinc ion [18]. By analogy it was suggested that the RING2 domain of Parkin may also bind just one zinc atom (Fig. 4A). However, this contrasts with the zinc titration analysis that suggested that Parkin can bind a total of eight zinc ions, two each for RING0, RING1, IBR, and RING2 [19].
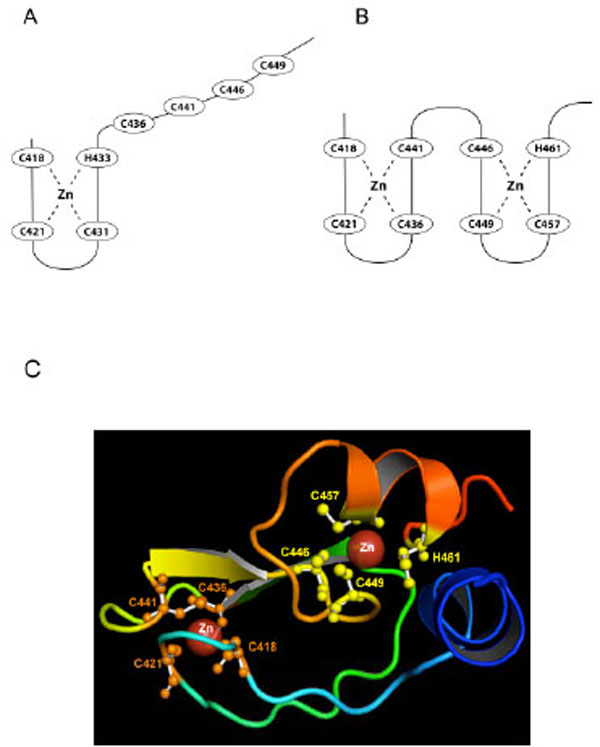
A) The previous RING2 model based on the Morett and Bork (1999) selection of ligands [104]. The last four Cys ligands are constrained in their ability to form a zinc ion binding site and it was suggested that Parkin RING2 had only one bound zinc ion. However, experimental evidence suggested that Parkin RING2 binds two zinc ions [19]. B) Our newly predicted structural model explains how two zinc ions may be bound by RING2. C) Our predicted model for the RING2 domain with ligand residues in the zinc binding sites shown in orange (first binding site) and yellow (second binding site).
While analyzing the predicted models of RING2 and the alignment with identified template proteins (RING2 of HHARI and IBR domain of RING finger domain of protein 31), we found that another conformation was possible for Parkin RING2 that uses a different set of ligands to accommodate two zinc ions. This model suggests that C418, C421, C436 and C441 coordinate to the first zinc ion while C446, C449, C457 and H461 coordinate to the second zinc ion (Fig. 4B, 4C). The zinc coordination sites in the predicted model are similar to the IBR domain of RING finger protein 31, which also binds two zinc ions. With regard to spacing, the previous model of RING2 follows the Lovering and Freemont RING spacing, except for the presence of 4 rather than 2 amino acids between ligand 5 and 6 (Fig. 2B). Our predicted structure follows the Lovering and Freemont RING spacing except there are 4 instead of 2 amino acid residues between ligand 4 and 5, and 3 rather than 2 residues between ligand 7 and 8 (Fig. 4C). A major point of difference is that in our predicted structure, the RING2 domain boundary, defined as being 4 residues before the first Cys and 6 residues after the last ligand (a histidine rather than cysteine in our predicted structure) is followed by only 4 amino acid residues rather than the 6 residues, as defined by Freemont [103]. The predicted RING2 structure does not form the classical cross brace structure and the last amino acid involved in zinc binding is His461 (Fig. 4B). Therefore our predicted structure varies in spacing, in the RING domain boundary, and also in location of the histidine. Wong et al. (2007) [21] had earlier proposed a similar first zinc ion ligand binding site (C418, C421, C436 and C441) for RING2 based on homology modeling.
Our RING2 structural model can explain several previously puzzling observations about Parkin. One is the number of zinc ions bound by Parkin,. Our model predicts two for RING2 for a total of eight in full length Parkin as demonstrated by Hristova et al. (2009) [19]. Another is the observed instability of Parkin C-terminal deletions and chimeric Parkin-HHARI proteins [29, 30]. In our predicted RING2 structure the deletion resulting from the W453X mutation would delete the final two ligands, C457 and H461 of the second zinc binding domain, explaining why this mutant spontaneously adopted an unfolded conformation [29]. Further, any C-terminal deletion greater than three amino acids resulted in the loss of Parkin solubility [29]. While H461 is the last ligand and fifth residue from the C-terminal, the importance of W462 in Parkin has remained largely unknown. The role of the highly conserved tryptophan residues in RING structures, e.g. Parkin W453 and W462, have been thought to be important for making interactions with other proteins, however the new insight into the metal-ion-recognition property of the indole ring of tryptophan suggests that the aromatic rings of these residues can form non-covalent interactions with zinc ions to provide rigidity to the RING structure and/or cause conformational changes leading to binding of zinc ions [106]. Therefore, as shown by Winkelhofer et al. (2003) [29] the last three residues, F463, D464, and V465, are the only dispensable C-terminal amino acids in Parkin RING2.
In contrast to Parkin, HHARI tolerated C-terminal deletions [30]. To address this difference, chimeras of the two proteins were prepared in which C-terminal fragments of HHARI from residues 376, 379 or 395 onwards, were substituted for Parkin residues 449 or 453 onwards. The chimeras were assayed for stability in situ [30]. In all of the chimeric proteins, the final two putative zinc ligands (C457 and H461) in our predictd RING2 model were substituted by amino acids G, Y, H, W, K or D. In one case, a His residue replaced the C457 ligand, however this construct was still missing the zinc binding ligand at position 461. All of the chimeras proved to be unstable, supporting the hypothesis that Parkin RING2 contains two zinc binding sites, and, as predicted by the model, both are important for Parkin stability.
Our RING2 model also explains the unexpected result regarding the behavior of the C431A mutation in an ambitious study in which the Parkin cysteines were individually mutated to determine which ones affected Parkin solubility [21]. The C431A mutation remained soluble, which was puzzling as all other cysteine mutants resulted in insoluble Parkin. Although C431 is considered as a ligand in the first zinc binding site in the Morett and Bork RING2 structure, C431 is not a ligand in either of the two zinc binding sites of our predicted RING2 structure (Compare Fig. 4A with 4B). This provides further support for the predicted RING2 structure.
Another previously puzzling observation came from Hampe et al. (2006) [27] in which mutation of either C418 or C441 in Parkin resulted in loss of autoubiquitination activity. Using the Morett and Bork (1999) ligand selection, C418 was a part of the first zinc binding site, whereas C441 was in the second zinc binding site. If Parkin RING2 was similar to HHARI RING2, we would expect the second zinc site to be unimportant for catalytic activity and hence the mutation of C441 would retain activity. However, that is not the result obtained by Hampe et al. (2006). Using our predicted RING2 structure, ligand C441 is a member of the first zinc binding site and therefore, like C418, should inhibit Parkin ubiquitination activity when mutated. This is consistent with the Hampe et al. (2006) [27] results.
From observations of zinc binding sites in RING domains discussed in Kentsis et al. [107], it appeared that zinc binding sites may show negative cooperativity in zinc binding, i.e. binding of zinc in one binding site reduces the affinity for binding of zinc to a second site. Considering that the new conformational structure proposed for the RING2 domain would explain several earlier observations, we hypothesize that Parkin assumes this conformation, at least part of the time. Whether or not the two zinc binding sites remain occupied continually, or whether Parkin can function (or remain soluble) with just one zinc ion bound remains to be determined. Indeed, domains with just one bound zinc ion have been shown to self-assemble into stable multi-RING structures that enhance the function of individual RING domains [107]. Based on these and related observations [108-110], an attractive possibility is that, in response to changing physiological conditions, a transition between bound and unbound zinc ions at one of the two zinc binding sites of Parkin RING2 promotes a change in local structure that acts as a mechanical switch, providing a mechanism whereby Parkin-protein association may be altered resulting in a change in function. Although an interesting hypothesis, a significant amount of work remains to be done with Parkin RING2 in exploring that hypothesis.
ACKNOWLEDGEMENTS
We thank Gil Ortiz , Graphic Designer Specialist for his expert assistance in preparing the line drawings representing the structure of the Parkin RING domains.
ABBREVIATIONS
REFERENCES
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link] [PMC Link]

