All published articles of this journal are available on ScienceDirect.

Molecules Acting on CB1 Receptor and their Effects on Morphine Withdrawal In Vitro
Abstract
Several pharmacological studies indicate that CB1 cannabinoid receptors (CB1Rs) are present in guinea pig ileum (GPI) and their activation reduce the acetylcholine (Ach) release. Dependence can be induced and measured in vitro by using GPI and the contraction due to opioid withdrawal is caused by acetylcholine release.
Design of molecules acting on the CB1Rs are widely studied and the large availaibility of CB1Rs agonists and antagonists provides powerful tools to determine the role of these receptors in mediating some of physiological and pharmacological effects in the myenteric neurones.
Given the relationship between CB1Rs/Opioid Withdrawal/Ach system, in the present paper we have designed six new CB1Rs agonists named A-F and evaluated their role in mediating morphine withdrawal in GPI. Also, a comparative study was performed by using the CB1Rs synthetic cannabinoid WIN 55,212-2 and CP 55,940. The results of our experiments indicate that both WIN 55,212-2 and CP 55,940 (1x10-8-5x10-8-1x10-7 M) were able to reduce morphine withdrawal in a concentration-dependent manner. Very similar results were obtained with the new CB1Rs agonists (A-F) used at same concentrations. The results of our experiments indicate that CB1Rs are involved in the control of morphine withdrawal in vitro thus confirming an important functional interaction between the cannabinoid and opioid system.
INTRODUCTION
Cannabinoid drugs exert a wide range of biological effects and are currently under study for their multiple potential therapeutic uses [1-4].
Cannabinoids and opioid share several pharmacological properties [5-7] and a strong interaction between opioid and cannabinoid systems has been reported [8-12]. This interaction can be studied by using GPI where cannabinoid CB1Rs have been found [13, 14]. The enteric nervous system has been considered as a simplified version of the central nervous system, considering its complex network-like organization and the presence of a large number of neurotrasmitters and neuromodulators. Isolated preparations, GPI, have been widely employed for assessing the acute effects of opioid and as a model for studying the interactions of opioid with other neuronal systems.
Dependence can be induced and measured in vitro by using GPI [15-18]. Tissues from untreated animals, after a brief exposure to opioids, show a strong naloxone-induced contracture [15-18] indicating that the cellular mechanisms of dependence may occur very rapidly following occupation of receptors and that these mechanisms operate within the myenteric plexus.
The characteristics of dependence development and the precipitation of withdrawal by naloxone in the guinea-pig ileum are very similar to those of acute dependence in experimental animals and man [15-18].
It has been demonstrated that Ach plays an important role in expression of opioid withdrawal because cholinergic agonist exacerbate opioid withdrawal; whereas, muscarinic and nicotinic blockers attenuate some aspects of the syndrome [19, 20]. Furthermore, a large proportion of the contraction due to opioid withdrawal is caused by acetylcholine release since it can be blocked by atropine or hyoscine [21, 22].
Several pharmacological evidences suggest that CB1Rs are present in the GPI and the effects on gastrointestinal motility depend on their activation which cause a reduction of Ach release [23-27].
Design of molecules acting on the CB1Rs are widely studied and the large availaibility of CB1Rs agonists and antagonists [28, 29] provides powerful tools to determine the role of these receptors in mediating some of physiological and pharmacological effects in the myenteric neurones.
Given the relationship between CB1Rs/Opioid Withdrawal/Ach system, in the present paper we have designed new CB1Rs agonists named A-F [30, 31] and evaluated their role in mediating morphine withdrawal in GPI. Also, a comparative study was performed by using the CB1Rs synthetic cannabinoid WIN 55,212-2 and CP 55,940.
MATERIALS AND METHODS
Morphine Withdrawal on Guinea-Pig Ileum
Male Charles River guinea-pigs (180-200 g) were used for all the experiments. Animal Care and use followed the directions of the Council of the European Communities (1986). The animals were housed in colony cage (4 guinea-pig each) under conditions of standard light (light on from 7.00 a.m. to 7.00 p.m.), temperature (22+1°C) and room humidity (60%+10%) conditions for at least 1 week before the experimental sessions. Food and water were available ad libitum.
The experimental procedure was that described previously [32]. The ilea were allowed to equilibrate for 40-60 min without washing and the response to acetylcholine (Ach) was determined for three times (10-6 M) so that response could be expressed as percentage of Ach maximum. A reproducible acute opiate dependence was obtained performing the following experimental procedure. A typical tracing of contracture responses of the ileum to repeated challenges with opiate and naloxone is shown in Fig. (1).
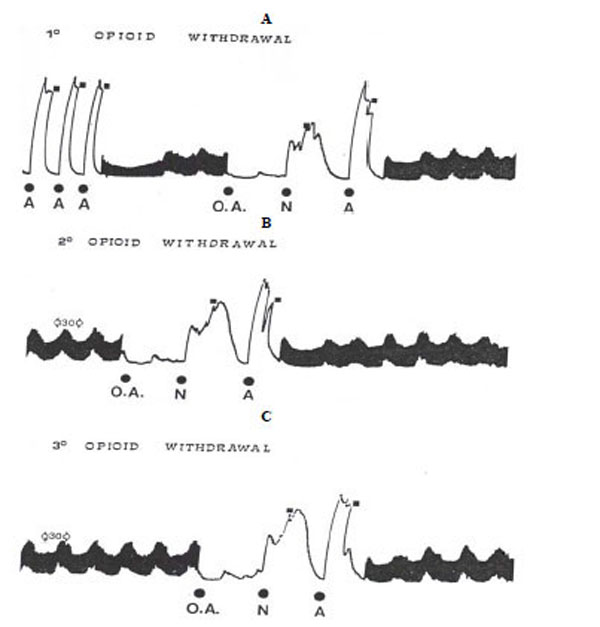
Typical tracing of opioid withdrawal on guinea-pig ileum. A. 3 similar acetylcholine response (A), electrical stimulation, injection of the opioid agonist (OA) followed after 4 min of contact period by naloxone (N) which induces contraction (1° opioid withdrawal). After washout (█), it was performed another A response.
B: After 30 min resting period under electrical stimulation, a further 4 min exposure of the ileum to the OA and N elicited reproducible response (2° opioid withdrawal).
C: After another 30 min resting period under electrical stimulation, the ileum responded again to the OA and N with the same intensity (3° opioid withdrawal).
After three similar Ach responses, the preparation was electrically stimulated for 10-20 min, (0.5 msec pulse delivered transmurally, at a frequency of 10 sec at supramaximal voltage, 25V). Before the addition of the morphine to the bath, the electrical stimulation was switched off. Under these conditions, the first contact with the opioid agonist followed after a 4 min exposure by naloxone induced a strong contraction (about 80% of the Ach maximum).
However, after washout, another Ach response was performed (to verify whether the ileum responsiveness was modified after withdrawal contracture) (Fig. 1A) and, after 30 min resting period under stimulation, a further 4 min exposure of the ileum (without electrical stimulation) to the opiate and naloxone elicited reproducible response.
Following washout, Ach response (Fig. 1B) and another 30 min resting period under stimulation, the ileum responded again to the morphine and naloxone with the same intensity (Fig. 1C). In our experiments, to avoid a possible tolerance for repeated morphine injection, each preparation was submitted only to three challenges with morphine and naloxone. Naloxone per se did not produce effects on “naive” preparations or those washed after opiate contact.
Experimental Procedure
The administration of CB1Rs agonists WIN 55,212-2, CP 55,940 and A-F was performed according to the following schedule:
- three Ach response
- electrical stimulation (10-20 min)
- morphine injection (10-5 M) in absence of electrical stimulation (4 min) and the addition of naloxone (10-5 M) with subsequent contraction (1° opioid withdrawal)
- washout and Ach response
- electrical stimulation (30 min)
- CB1Rs agonists WIN 55,212-2, CP 55,940 or A-F (1x10-8-5x10-8-1x10-7 M) without electrical stimulation injected 10 min before morphine followed by naloxone (2° opioid withdrawal)
- washout Ach response
- electrical stimulation (30 min)
- final control opiate withdrawal (3° opioid withdrawal)
Drugs
Naloxone HCl and WIN 55,212-2 (R(+)-(2,3-dihydro-5methyl-3-((morpholinyl)methyl)pirrolo-(1.2.3.-sw)-1.4-benzoxazinyl)-(1-naphthalenyl)methanone)mesylate, GDP and GTPγS were purchased from Sigma Chemical Co. (St. Louis, MO, USA); morphine HCl was from Carlo Erba (Milan, Italy), CP 55,940 was obtained from Tocris (Bristol, UK). Rat brains were purchased from Pelfreeze Rogers, AR. [35S]GTPγS was purchased from New England Nuclear Corp., Boston, MA.
Parameter Evaluation
Four parameters were evaluated:
- Naloxone contracture: the size of the contraction produced by the naloxone challenge was expressed as a fraction of the maximum contraction obtained with the subsequent addition of Ach in the same piece of tissue according to the method previously described [32]:
- Ach response before and after the treatment: reduction or increase of the Ach responses in the post-drug expressed as a percentage of Ach response in the pre-drug.
- Electrically stimulation contraction before and after the treatment: reduction or increase of the electrically stimulation contraction in the post-drug was expressed as a percentage of the electrical stimulation in the pre-drug.
- Naloxone contraction before and after treatment: reduction or increase of the naloxone contraction in the post-drug was expressed as a percentage of the naloxone contraction in the pre-drug.
Statistical Analysis
Results were tested for statistical significance using the Student's t-test for paired data when results before and after treatments on the same preparation were compared. The ED50 were computed from the dose-response curve by the method of Litchfield and Wilcoxon [33].
Chemistry Pathway
The CB1Rs (A-F) were synthesized as described previously [30,31]. Table 1 shows the structures of the new CB1Rs agonists (A-F).
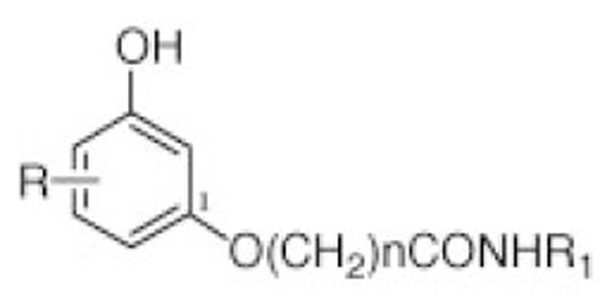
| CB1 Analogues | R | n | R1 |
|---|---|---|---|
| CB1R-A | 5-n-phenyl | 5 | CH2CH2OH |
| CB1R-B | 5-n-phenyl | 5 | c.C3H5 |
| CB1R-C | 5-n-phenyl | 10 | CH2-c-C3H5 |
| CB1R-D | 5-n-phenyl | 10 | CH2CH2OH |
| CB1R-E | 5-n-phenyl | 7 | CH2CH2OH |
| CB1R-F | 5-n-phenil | 7 | c.C3H5 |
Binding Assay
1. [35S]GTPγS Binding Assay. Cerebellar Membrane Preparation
The procedure was adapted from the method of Dodd et al. [34] The stripped rat brains were slightly thawed, and using a spatula, the cerebellum was removed and discarded; the remaining tissue was homogenized in ice-cold homogenization buffer (0.32 M sucrose, 10 mM Tris, 5 mM EDTA, pH 7.4). The homogenate suspension was centrifuged at 3700g for 10 min. The supernatant was decanted, and 12 mL was layered over 10 mL of 1.2 M sucrose. These tubes were centrifuged in a L7-65 ultracentrifuge using a 50.2 Ti rotor at 4 ˚C for 29 min at 44 000 rpm. The layer at the interface was then removed and subjected to a second sucrose spin over 0.8 M sucrose. The pellet was resuspended in TME buffer (25 mM Tris base, 5 mM MgCl2, 1 mM EDTA, pH 7.4), aliquoted, and stored at -70 ˚C. Protein was determined using the method of Markwell et al. [35].
2. [35S]GTPγS Binding Assay (Table 2)
EC50 and Emax Values of Anandamide Analogues for Stimulating [35S]GTPγS Binding in Rat Microsomal Membranes a
| CB1 Analogues | EC50 (µM) | Emax (%) |
|---|---|---|
| A | 0.48 ± 0.03 | 112 ± 4.8 |
| B | 0.22 ± 0.07 | 121 ± 1.5 |
| C | 0.34 ± 0.08 | 133 ± 3.5 |
| D | 0.52 ± 0.05 | 108 ± 2.7 |
| E | 0.42 ± 0.03 | 103 ± 1.9 |
| F | 0.27 ± 0.04 | 127 ± 4.5 |
| WIN 55,212-2 | 0.18 ± 0.09 | 144 ± 6.5 |
| CP-55,940 | 0.10 ± 0.05 | 135 ± 9.0 |
a [35S]GTPγ-S binding assay was conducted using eight concentrations of each analogue being tested and two experiments run with four duplicates of each point.
Our assay was based on a method by Selley et al. [36] and was adjusted for a 96-well plate analysis. Briefly, the rat membrane preparation (40-50 µg of protein) was incubated for 1 h at 30 ˚C in assay buffer (10 mM Tris, 100 mM NaCl, 5 mM MgCl2, 0.1% BSA) with 50 µL of 50 µM GDP, 50 µL of 0.05 nM [35S]GTPγS, or 100 µL of either; 10 µM GTPγS was used to measure the nonspecific binding, a series of different concentrations of the analogues being tested, or buffer alone as a control to obtain the baseline of GTPγS stimulation. The reaction was terminated by rapid filtration through Whatman GF/B filters, with ice-cold wash buffer containing 0.5% bovine serum albumin using the Packard Filtermate. Bound radioactivity was measured on the Packard Top-Count microplate scintillation counter.
RESULTS
Effect of WIN 55,212-2, CP 55,940 or CB1R Agonists (A-F) on Morphine Withdrawal
Both WIN 55,212-2 and CP 55,940 (CB1Rs synthetic cannabinoid) at 1x10-8-5x10-8-1x10-7 M were able to
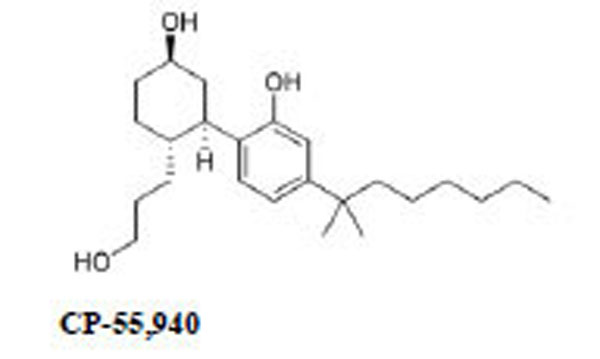
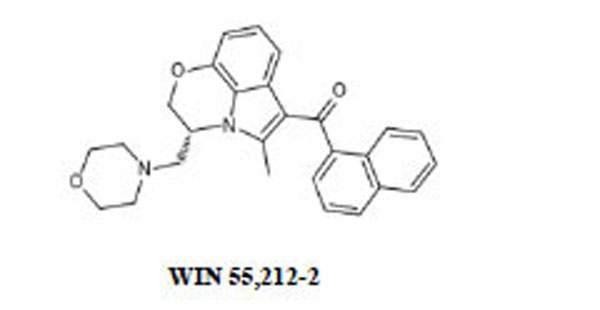
respectively prevent or reverse the naloxone-induced contraction in GPI exposed to morphine (Table 3).
ED50 and 95% C.L. Values of CB1Rs Agonist WIN 55,212-2, CP 55,940 or A-F (1x10-8-5x10-8-1x10-7 M) on Morphine Withdrawal. Each CB1Rs Agonist was Administered 10 min Before (A) or After (B) Morphine
| Compounds | Morphine Withdrawal (A) | Morphine Withdrawal (B) |
|---|---|---|
| WIN 55,212-2 | 2.1x10-8 M (1.5x10-9-2.4x10-7) | 3.1x10-8 M (5.5x10-9-3.1x10-7). |
| CP 55,940 | 2,5x10-8 M (3.5x10-9-4.3x10-7) | 3.5x10-8 M (2.7x10-9-4.3x10-7). |
| CB1R-A | 3.5x10-8 M (2.5x10-9-1.9x10-8) | 3.7x10-8 M (4.5x10-9-2.3x10-7) |
| CB1R-B | 2.7x10-8 M (3.4x10-9-1.7x10-8) | 2.9x10-8 M (3.5x10-9-1.7x10-7) |
| CB1R-C | 3.1x10-8 M (2.9x10-9-4.1x10-8) | 3.5x10-8 M (2.5x10-9-4.3x10-7) |
| CB1R-D | 2.9x10-8 M (1.3x10-9-3.3x10-8) | 2.2x10-8 M (1.5x10-9-3.3x10-7) |
| CB1R-E | 2.4x10-8 M (3.3x10-9-1.9x10-8) | 3.2x10-8 M (4.5x10-9-2.3x10-7) |
| CB1R-F | 2.5x10-8 M (3.3x10-9-1.6x10-8) | 3.6x10-8 M (4.5x10-9-2.3x10-7) |
Also, six new CB1Rs agonists (A-F) (1x10-8-5x10-8-1x10-7 M) were able to reduce morphine withdrawal (Table 3).
Electrical stimulation was also reduced by the above CB1 receptor agonists (Table 4), the final morphine withdrawal was still reduced (Table 4), whereas the Ach response was not modified (Data not shown).
The Effect of CB1Rs Agonist WIN 55,212-2, CP 55,940 or A-F [(1) 1×10-7-(2) 5×10-8- (3) 1×10-8 M] on Electrical Stimulation and Final Opioid Withdrawal
| Compounds | Electrical Stimulation | Final Morphine Withdrawal |
|---|---|---|
|
|
||
| WIN 55,212-2 | 75.6±3.9** 1 | 62.5±5.3** 1 |
| 46.8±2.7** 2 | 39.7±3.1* 3 | |
| 35.4±1.6** 3 | 22.4±2.3 3 | |
|
|
||
| CP 55,940 | 82.3±4.8** 1 | 57.3±2.5** 1 |
| 56.3±2.4** 2 | 44.8±4.9* 2 | |
| 37.3±2.7 3 | 35.3±2.4 3 | |
|
|
||
| CB1R-A | 72.8±2.5** 1 | 72.3±3.7** 1 |
| 49.7±2.9** 2 | 57.2±3.6** 2 | |
| 33.2±3.6 3 | 41.5±3.2* 3 | |
|
|
||
| CB1R-B | 77.5±4.2** 1 | 69.5±4.6** 1 |
| 59.3±3.6** 2 | 42.5±2.3** 2 | |
| 23.7±2.5 3 | 28.7±3.9 3 | |
|
|
||
| CB1R-C | 69.3±5.8** 1 | 68.7±5.1** 1 |
| 48.4±3.2** 2 | 44.3±2.1** 2 | |
| 32.3±2.1 3 | 20.7.6±1.9 3 | |
|
|
||
| CB1R-D | 73.1±6.7** 1 | 70.2±6.2** 1 |
| 55.1±3.2** 2 | 48.2±3.3** 2 | |
| 38.3±2.5 3 | 27.3±3.1 3 | |
|
|
||
| CB1R-E | 67.3±6.1** 1 | 73.4±4.3** 1 |
| 46.9±2.9** 2 | 62.5±2.5** 2 | |
| 36.2±6.5 3 | 44.3±5.8 3 | |
|
|
||
| CB1R-F | 80.1±5.7** 1 | 79.2±6.1** 1 |
| 63.9±3.5** 2 | 59.3±3.1** 2 | |
| 49.2±3.9** 3 | 46.2±2.2** 3 | |
Results are expressed as % of inhibition (mean±s.e.m.);
* P<0.05;
** P<0.01. All data were compared to the pre-drug.
DISCUSSION
While there are several data in literature on the effects exerted by cannabinoids on several effects induced by opioids [37-40], this is the first paper which evaluates the role of CB1Rs agonists in mediating morphine withdrawal in vitro. Furthermore, a comparative study was also performed by using both the well known CB1Rs agonists WIN 55,212-2 and CP 55,940 (CB1 synthetic cannabinoid) and six new CB1Rs agonists (A-F). The results of the present study indicate that both the synthetic cannabinoid WIN 55,212-2 or CP 55,940 as well as the six new CB1Rs agonists (A-F) were able to produce significant reduction on morphine withdrawal in vitro thus confirming the important functional interaction between the cannabinoid and opioid system.
However it is of interest to note that CB1Rs agonists (A-F) compared to the synthetic cannabinoid WIN 55,212-2 and CP 55,940 showed a very similar activity in inhibiting morphine withdrawal confirming that CB1Rs agonists (A-F) show a CB1 receptor affinity very similar to WIN 55,212-2 and CP 55,940 as shown in Table 2.
The discussion on the possible mechanism by which CB1Rs agonists causes a reducing effect on morphine withdrawal is open and several possibilities may be considered.
Ach system has been widely implicated in many of the pharmacological effects of opioids. Manipulation that alter the activity of Ach in the central nervous system frequently modify the effects of morphine and other opioid drugs [41-44].
Several authors have demonstrated that Ach agonists and antagonists are able to influence opiate withdrawal in vitro, suggesting an important functional interaction between the Ach system and opioid withdrawal [19-22]. Cholinergic agonist exacerbate opioid withdrawal; whereas, muscarinic and nicotinic blockers attenuate some aspects of the syndrome [19-22]. Furthermore, a large proportion of the contraction due to opioid withdrawal is caused by Ach release since it can be blocked by atropine or hyoscine [19-22]. Also, it has been demonstrated that also selective muscarinic receptors antagonists are able to influence opioid withdrawal in vitro [45].
Several pharmacological evidences suggest that CB1Rs are present in the myenteric neurones and the effects on gastrointestinal motility depend on the activation of CB1Rs which induce a reduction of Ach release [23-27]. Therefore, the ability of CB1Rs agonists to reduce morphine withdrawal may be related to their ability to block the release of Ach at presynaptic level [23-27] and confirming the role played by Ach in the expression of opiate withdrawal [19-22].
Furthermore, in our experimental conditions, the ability of CB1Rs agonists to reduce electrical stimulation after washout without altering the Ach response at postsynaptic level confirm a direct action on presynaptic Ach receptors [23-27].
Therefore, our data confirm and extend previous data [19-22] indicating a significative role of the cannabinoid system in the development of morphine withdrawal indicating that CB1Rs play an important role in the control of morphine withdrawal probably through Ach system [23, 27, 46].
However, another possibility should be considered.
The effects of CB1Rs on morphine withdrawal may be also related to their ability to block the release of adrenaline or dopamine by pre-synaptic receptors. Recently, it has been demonstrated that catecolaminergic system is involved in the control of the expression of opioid acute dependence in GPI [32, 47]. Several studies indicate that CB1Rs are able to block the release of dopamine and noradrenaline at presynaptic level of guinea pig ileum electrically stimulated [48]. Therefore, we cannot exclude the possibility that CB1Rs reduce morphine withdrawal also by blocking the release of catecolamines by the presynaptic neurons.
Regarding the comparative study performed between the well known CB1Rs WIN 55,212-2 and CP 55,940 (CB1 synthetic cannabinoid) and the six new CB1Rs agonists (A-F). our data show a good correlation between the binding activity and biological activity,. Therefore, our results indicate that the aromatic structure, the length of aliphatic chain and the chain of 5-10 carbon atoms assure the affinity of compounds to CB1Rs.
Finally, the possible implications of this finding in the context of novel therapeutic aspect may be considered suggesting that CB1Rs activation or inhibition may result benefical as additional therapeutic strategy during opioid withdrawal.
REFERENCES
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link] [PMC Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]

