All published articles of this journal are available on ScienceDirect.

The Role of Exercise on L-Arginine Nitric Oxide Pathway in Chronic Heart Failure
Authors Info & Affiliations
Abstract
Chronic heart failure (CHF) is a pathological state with high morbidity and mortality and the full understanding of its genesis remain to be elucidated. In this syndrome, a cascade of neurohormonal and hemodynamic mechanisms, as well as inflammatory mediators, are activated to improve the impaired cardiac function. Clinical and experimental observations have shown that CHF is associated with a generalized disturbance in endothelium-dependent vasodilation, which may contribute to the progression of ventricular and vascular remodelling in this syndrome. There is also accumulating evidence that disturbances in nitric oxide (NO) availability is involved in the development of heart failure at the systemic and cardiac levels. NO is a ubiquitous signalling molecule which causes potent vasodilation, inhibits platelet activation and regulates the contractile properties of cardiac myocytes. It is generated from the amino acid L-arginine via constitutive and inducible isoforms of the enzyme NO synthase (NOS). There is evidence that exercise, a nonpharmacological tool, improves symptoms, fitness (VO2peak), quality of life and NO bioavailability in CHF population. This review examines different aspects of the L-arginine-NO pathway and inflammation in the physiopathology of CHF and highlights the important beneficial effects of exercise in this disease.
1. INTRODUCTION
Chronic heart failure (CHF) is a clinical syndrome caused by an abnormality of the heart and diagnosed by a characteristic patterns of haemodynamic, renal and hormonal responses [1]. The prevalence of this disease is high throughout the world, with 50% of patients dying within four years and of 50% of patients with severe heart failure dying within one year [2].
Endothelium-dependent dilation of resistance vessels in response to acetylcholine is impaired in heart failure and related to the clinical severity of the disease [3, 4]. Endothelial dysfunction seems to contribute to the high vascular resistance in the systemic and pulmonary vascular beds of patients with chronic heart failure [3-5]. Accumulating evidence suggests that endothelial dysfunction may result from a decreased release of nitric oxide (NO) or an enhanced inactivation of this bioactive mediator [6].
In mammalian cells, NO is formed from the terminal guanidino nitrogen atom of L-arginine by a family of NOS enzymes with characteristics of cytochrome P-450 reductase [7-9]. The principal mechanism of action of NO is to interact with heme moiety of guanylyl cyclase, increasing the production of intracellular cyclic guanosine monophosphate (cGMP) [9, 10]. Membrane transport of the precursor L-arginine is essential for physiological NO production and alterations in transport may in part help explain several of the pathological changes observed in CHF [6].
Increasing evidence has demonstrated that habitual physical activity can reduce the risk of cardiovascular disease [11]. Indeed, a direct correlation of the amount of exercise performed and a reduction in mortality due to cardiovascular disease has been reported in middle-aged and elderly populations [12].
This review aims to provide an overview of clinical and experimental data on the role of L-arginine-nitric oxide pathway and inflammation in the pathophysiology of CHF and the potential beneficial effects of exercise in this disease.
2. PATHOPHYSIOLOGY OF CHRONIC HEART FAILURE
CHF is a common and complex clinical syndrome whose incidence and prevalence is increasing world-wide [13]. CHF results from structural or functional cardiac disorders, leading to an impaired ability of the ventricle to fill with or eject blood. The cardinal manifestations of CHF are dyspnea and fatigue, which may limit exercise tolerance, and fluid retention, which may lead to pulmonary congestion and peripheral edema [1]. Both abnormalities can impair the functional capacity and quality of life of affected individuals, but they do not necessarily dominate the clinical picture at the same time. Some patients have exercise intolerance but little evidence of fluid retention, whereas others complain primarily of edema and report few symptoms of dyspnea or fatigue [2, 14].
Abnormal left ventricular systolic function from any cause appears to stimulate compensatory mechanisms to maintain cardiac output, systemic blood pressure and perfusion to vital organs. Most important among these are: (1) the Frank-Starling mechanism (i.e. the ventricle responds to an increased preload by enhancing its contraction; (2) ventricular hypertrophy with or without chamber dilation; and (3) neurohormonal activation [13-15].
Many neurohormonal systems are activated during chronic heart failure, such as the renin-angiotensin-aldosterone system, sympathetic nervous system, endothelin and vasopressin, which lead to vasoconstriction and water and sodium retention [13, 15, 16]. Neuroendocrine systems are interactive and integrated with the brain, kidneys, heart and periphery, playing a role in the onset and progression of heart failure. Despite presumed initial circulatory homeostasis, prolonged neurohormonal activation may become highly maladaptive, contributing not only to increased afterload and reduced cardiac output but also causing necrosis and death of myocardial cells [13, 15, 16].
The kidney plays a central role in salt and water retention in heart failure. A significant reduction in glomerular filtration rate accompanies impaired cardiac output and is caused by reduced arterial pressure and by constriction of the preglomerular arterioles in response to increased sympathetic activity and angiotensin II production [13, 15, 17]. This reduction in the glomerular filtration rate reduces sodium delivery to the nephron and, associated with direct actions of angiotensin II on tubular sodium reabsorption, may cause sodium and water retention [17, 18]. The kidney synthesises rennin, protease kallikrein and prostaglandins, which are increased during chronic heart failure [19, 20].
Whereas neurohormonal factors have been extensively studied, the impact of local factors regulating peripheral vasomotor tone and tissue perfusion in chronic heart failure has been poorly defined. Clinical and experimental observations have demonstrated that chronic heart failure is associated with a generalised impairment of endothelium-dependent vasodilation, which may contribute to the progression of ventricular and vascular remodelling in this syndrome (Mendes Ribeiro et al. 2001).
3. L-ARGININE – NITRIC OXIDE PATHWAY
In 1980, Furchgott et al. (1980) first reported that endothelial cells release an endothelium-derived relaxing factor (EDRF). In 1987, this molecule was identified as being NO [21, 22], a labile gas that can diffuse readily across membranes and with a half-life ranging from 2 to 30 s. NO can be reduced by the presence of molecular oxygen (O2), superoxide anions (O2-●) and heme proteins [23-25]. In mammalian cells, NO is synthesized from the terminal guanidino nitrogen atoms of L-arginine, a cationic amino acid, by NO synthases (NOS) (Fig. 1). This reaction involves two monooxygenation steps requiring O2 and NADPH: an initial hydroxylation of L-arginine to generate N-hydroxy-L-arginine, followed by oxidation which leads to the formation of NO and L-citrulline [26, 27].
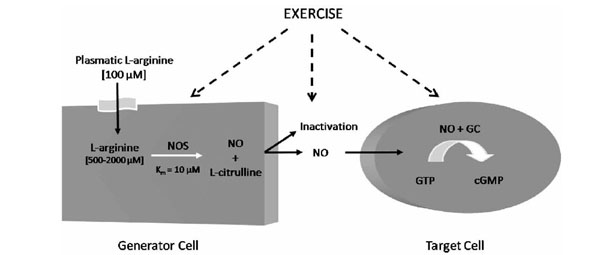
L-Arginine Nitric Oxide Pathway. GC: guanylyl cyclase; cGMP: cyclic guanosine monophosphate; NO: Nitric oxide; NOS: Nitric oxide; synthase.
To date, three different isoforms of NOS have been identified: type I or neuronal (nNOS), type II or inducible (iNOS) and type III or endothelial (eNOS) [9, 26]. The isoforms nNOS and eNOS are constitutive and their activities are regulated by intracellular free calcium and the calcium-binding protein calmodulin, whereas the iNOS is calcium independent. Despite the well established role of the three NOS isoforms, recent evidence support the idea of NO production via the mitochondrial membrane in different cell types such as liver, kidney and cardiac cells [28-31]. This isoform is known as mitochondrial NOS (mtNOS) and it is still unclear if mtNOS is a new isoform of NOS or a splicing of nNOS [30, 32, 33].
Approximately 10 nM of NO is needed to activate guanylyl cyclase (GC)-coupled receptors leading to cGMP accumulation in vascular smooth muscle cells [34, 35]. Studies in vivo suggested that GC will display a dual regulation by NO: long-lasting tonic activity due to persistent occupation by NO of the heme binding site and phasic activity due to engagement of another unidentified site [34]. The cGMP influences a number of vascular cell types and regulates vasomotor tone, endothelial permeability, cell growth and differentiation, as well as platelet and blood cell interactions [36]. Moreover, cGMP increases the expression of methylarginine dimethylamino-hydrolase (DDAH), an enzyme that metabolizes asymmetric dimethyl-arginine, N(G)-dimethyl-L-arginine (ADMA) and N(G)-monomethyl-L-arginine (L-NMMA), which are competitive endogenous inhibitors of NOS. It is possible that this effect serves to maintain NO levels in the endothelial environment [37].
L-arginine is considered a semi-essential amino acid in most adult mammals, but under certain conditions where the synthesis of L-arginine is decreased and/or its catabolism is increased, L-arginine becomes an essential amino acid [38]. In humans, the kidney and dietary intake are responsible for maintaining normal plasma levels of this amino acid, which range from 80 to 100 μM in healthy patients [39, 40].
Membrane transport of L-arginine in mammalian cells is predominantly mediated via four transport systems: y+, y+L, Bo,+ and bo,+, with different kinetic patterns [41, 42]. Several studies have demonstrated that L-arginine transport is rate limiting for generating NO by endothelial cells activated macrophages, platelets, endothelium and brain astrocytes, despite the fact that the intracellular concentration of L-arginine remains well above Km values for NOS [43-47]. This special feature is known as the “L-arginine paradox” [48, 49]. There are also studies suggesting the participation of citrulline-arginine recycling via the citrulline-NO cycle in high-output production of NO in activated microglial cells and in endothelial cells [50].
4. L-ARGININE – NO PATHWAY IN CHRONIC HEART FAILURE
There is general consensus that the basic disturbance underlying the pathology of heart failure is the loss of functioning, necrotic or apoptotic myocardial cells, resulting in an inadequate blood flow to peripheral tissues [51, 52]. In this syndrome, a cascade of neurohormonal and hemodynamic mechanisms, such as activation of the sympathetic system and renin-angiotensin-aldosterone system, are activated to preserve cardiac function. However, prolonged activation of these pathways is deleterious and is responsible for the development of chronic heart failure [51, 53]. Furthermore, increased oxidative stress and the presence of an inflammatory status may contribute further to cardiac remodelling and vascular dysfunction observed in this illness [54-57]. CHF is thus a very complex syndrome characterised by a myriad of cellular and systemic alterations. There is accumulating evidence that the L-arginine-NO pathway has an important role in the pathophysiology of this disorder [6].
According to several studies, endothelial dysfunction could result from decreased release or enhanced inactivation of NO by increased oxidative stress found in CHF [3]. Therefore, disturbances of the L-arginine-NO pathway reported in CHF may have an important role in the pathogenesis of this syndrome (Table 1). Decreased activity of the L-arginine-NO pathway is suggested by reduced urinary excretion of nitrates in heart failure patients at rest and during exercise after infusion of L-[15N]-arginine and by decreased eNOS expression and NO synthesis in the aortic endothelium from experimental rodent models of heart failure [58-61]. Clinical and animal models of CHF have reported increased plasma levels of ADMA, which would inhibit NO production and lead to decreased endothelium-dependent relaxation [62-65]. Moreover, elevated levels of ADMA have been proposed as a predictor of adverse cardiovascular outcomes in patients with heart failure [66]. In contrast, several clinical studies have demonstrated that patients with heart failure exhibit an increased responsiveness to inhibitors of NO synthesis and elevated plasma levels of stable NO breakdown products [67-70], suggesting that NO synthesis is increased rather than decreased.
Modulation of the L-Arginine-NO Pathway in Chronic Heart Failure
| Reported Change | Summary of Findings | Reference | |
|---|---|---|---|
| NOS activity/ NOS expression/ nitrite+nitrate production /cGMP | Increased | ↑ plasma nitrate levels | Chowdhary, et al. 2002 |
| Increased | ↑ vasoconstrictor effect of L-NMMA | Dover et al. 2006 | |
| Decreased | L-arginine administration ↓ vascular resistance associated with ↑ nitrate/nitrite production | Katz et al. 1999 | |
| Increased | ↑ NO plasma concentration | Kaye et al. 1998 | |
| Increased | ↑ iNOS expression in skeletal muscle | Stein et al. 1998 | |
| Increased | ↑ iNOS expression and iNOS protein content in skeletal muscle inversely correlated with changes in COX-activity and the exercise | Gielen et al. 2005 | |
| Increased | ↑ endomyocardial eNOS or iNOS gene expression augments LV stroke volume and LV stroke work | Heymes et al. 1999 | |
| Decreased | ↓ activity of the L-arginine-NO metabolic pathway | Katz et al. 1999 | |
| Increased | iNOS, but not eNOS expression in ventricular myocardium | Drexler et al. 1999 | |
| Increased | ↑ plasma nitrite level | Yu et al. 2001 | |
| Increased | ↑ iNOS expression in the myocardium and ↑ serum concentration of nitrate/nitrite | Orús et al. 2000 | |
| Increased | ↑ NO plasmatic concentation which showed positive correlation with TNF-α plasmatic levels | Elahi et al. 2007 | |
| Increased/decreased | ↑ iNOS activity and expression, but ↓ eNOS activity and expression in failing hearts | Ferreiro et al. 2004 | |
| Unaltered | There was no difference in exhaled NO between controls and heart failure patients, nor after exercise | Seshadri et al. 2003 | |
| Endothelial function | Decreased | responses vasodilatador endothelium-dependent induce by acetylcholine in braquial artery | Morgan et al. 2004 |
| Decreased | ↓ forearm blood flow | Ishibashi et al. 2000 | |
| Decreased | ↓ vasodilatation endothelium-dependent in forearm | Heitzer et al. 2005 | |
| Increased | Vitamin C improves endothelium-dependent forearm vasodilatation | Hirooka et al. 2003 | |
| Decreased | ↓ endothelium-dependent vasodilatation in response to aceylcholine in internal radial artery was improved by oral L-arginine supplementation and exercise training. | Hambrecht et al. 2000 | |
| Decreased | ↓ forearm blood flow | Bank et al. 2000 | |
| Decreased | ↓ forearm blood flow in response to acetylcholine. | Sugamori et al. 2002 | |
| L-arginine transport | Increased | ↑ L-arginine transport via system y+ in erythrocytes from heart failure patients associated with ↓ plasmatic levels of L-arginine and ↑ plasmatic levels of L-NMMA | Hanssen et al. 1998 |
L-NMMA, NG-monomethyl L-arginine; NO, nitric oxide; NOS, nitric oxide synthase; iNOS, inducible nitric oxide synthase; eNOS, endothelial nitric oxide synthase; PBMCs, peripheral blood mononuclear cells; COX, cytochrome e oxidase.
Furthermore, an activation of L-arginine uptake in erythrocytes and aorta were found in patients and in rodent models of heart failure, respectively [71, 72]. According to these findings, it is possible that the increment in L-arginine transport could represent an adaptive response to increase L-arginine availability for a putative increase in basal NO production in heart failure. On the other hand, Kaye et al. [69] have shown an impairment of L-arginine uptake associated with the reduction of CAT-1 expression in myocardial cells from chronic heart failure patients.
Our group has detected the presence of a disturbance in the plasma amino acid profile in CHF, with concentrations of most amino acids lower compared to healthy age-matched controls [72]. The plasma levels of L-arginine in heart failure were reduced by 50% of control values (Table 1). In this context, increased L-arginine transport observed in erythro-cytes from CHF patients may reflect a widespread compensatory response to the decreased plasma L-arginine to sustain elevated production of NO. We have also report that plasma levels of L-NMMA are elevated in CHF patients [72].
The presence of nNOS, eNOS and iNOS have been detected in myocardium [73]. Increasing evidence indicates that expression and activity of iNOS is increased in different tissues and cells in heart failure [71, 74, 75]. In contrast, conflicting data exist with regard to eNOS expression and activity [73]. nNOS seems also to be involved in the pathogenesis of heart failure, and according to one study, myocardial cells from heart failure patients have an increased expression and activity of nNOS [76]. The reasons underlying conflicting findings in the L-arginine-NO pathway in chronic heart failure are unknown and may reflect the diverse causes and severity of this disease.
5. EFFECTS OF EXERCISE IN HEART FAILURE PATIENTS
It is well known that exercise training leads to beneficial adaptations in patients with heart failure, including an improvement in symptoms and quality of life, reduction of fatigue and dyspnea, and enhancement of exercise tolerance. Thus, exercise training is strongly recommended by the American Heart Association [14] and the European Society of Cardiology [2], as part of the treatment for mild to moderate heart failure. These patients usually have a very low exercise capacity with VO2 peak levels between 12 to 15 mL.kg-1.min-1, approximately half of that observed in healthy individuals matched by age and gender [77]. Improving exercise tolerance, and consequently VO2peak, is important for two major reasons: (1) the minimum threshold for independent living is 18 and 15 mL.kg-1.min-1 for elderly men and women, respectively [78]; and (2) heart failure patients with VO2peak lower than 14 mL.kg-1.min-1 are candidates for heart transplantation [79].
There is an extensive body of knowledge regarding the positive effects of exercise on functional capacity, demonstrated by an increase in VO2peak, an improvement in the New York Heart Association functional classification, quality of life and functional autonomy [80]. The benefits are not restricted to the functional aspect, but extend to a lower mortality rate and to hospital readmission [81]. It is worth noting, however, that more conclusive data will be available after the termination of the trial Heart Failure: A Controlled Trial Investigating Outcomes of Exercise Training (HF-ACTION). This is the largest trial investigating the effects of exercise on HF patients and has been conducted since 2002 in more than 50 U.S. and Canadian hospitals involving 3000 patients.
The causes of exercise intolerance and the precise mechanisms for the beneficial effects of exercise in heart failure are still being extensively discussed. It has been suggested that central (related to the heart) and peripheral (related to the muscle and the vasculature) factors, in addition to neurohormonal modifications, can contribute to exercise intolerance [82]. An in-depth review about the central, metabolic, and neurohormonal adaptations associated with exercise training is beyond the scope of this review.
Exercise has also been shown to improve some alterations in neuro-hormonal overactivity found in CHF. A reduction in norepinephrine spillover, enhancement in the vagal high frequency component of heart rate variability, and a decrease in the sympathetically mediated low-frequency oscillation have been demonstrated. [83, 84]. Although controversial, it is possible that these effects of exercise are secondary to skeletal muscle or peripheral vascular training improvement [83-85]. Recently, consistent data has suggested that reduction in sympathetic nerve activity and improvement in baroreflex function in CHF after exercise are due to the concomitant reduction in angiotensin II and angiotensin receptors in the central nervous system [86].
5.1. Vascular Adaptations to Exercise in Heart Failure Patients: Role of L-Arginine-NO Pathway
Exercise intolerance can also result from the systemic vasoconstriction and diminished peripheral perfusion commonly seen in heart failure. In this context, the endothelium, and especially NO, constitute central figures in vasomotor tone and perfusion regulation [87]. It has been consistently demonstrated that exercise training restores endothelium function in CHF patients. An increase in the brachial artery diameter [88] or in forearm blood flow [89] was observed after infusion of acetylcholine, but not by nitroglycerine or sodium nitroprussiate, indicating an improvement in endothelium-dependent and non–independent vasodilation. Flow-dependent vasodilation is also increased after exercise training [90]. These changes were observed after at least 4 weeks of different exercise modes, including aerobic training on a cycle ergometer [87], local muscular endurance training (handgrip) [89] and circuit training involving resistance plus aerobic training [91]. Table 2 presents a summary of the trials involving exercise and vascular changes in heart failure patients.
Effects of Exercise in Heart Failure Patients
| Subjects | Exercise | Results | Ref. |
|---|---|---|---|
| 52 M (55±10 yr) EF = 31±7% NYHA II / III |
8 wk. Aerobic 60% VO2peak 3 x 40 min |
↑ VO2peak Sig. correlation between changes in VO2peak and EDVD ↑ FDVD ↔ EIVD |
Belardinelli et al. 2006 |
| 12 (60±2 yr) NYHA I/II EF=26±3% |
8 wk Resistance (55-65% 1RM)+ aerobic 70-85% HRpeak 3 x 60 min |
↑ VO2peak ↑ EDVD ↑ EIVD |
Maiorana et al. 2000 |
| 10 M (55±4 yr) NYHA II/III EF=19±9% |
4 wk. Handgrip training 60 N 6x/day |
↑ EDVD ↑ FDVD ↔ EIVD |
Hambrecht et al. 2000 |
| 10 (54±4 yr) EF=24±4% NYHA II/III |
24 wk. Aerobic 5x40 min 70% VO2peak |
↑ VO2peak ↑ forearm BF (ACh) ↔ forearm BF (NTG) ↑ BF velocity ↑ inhibitory effect of L-NMMA on forearm BF |
Hambrecht et al. 1998 |
| 12 (10 M, 58±3 yr) EF=25±5% NYHA II/III |
8 wk. Handgrip 5x30 min 70% maximal work rate 20 cont/min |
↑ forearm BF (ACh) ↔ forearm BF (NTG) ↑ reactive forearm BF |
Katz et al. 1997 |
| 11 M (55±10 yr) EF = 26±3% NYHA II / III |
6x/day. Aerobic 10 min 70% VO2peak |
↑ VO2peak ↑ EDVD ↑ FDVD ↔ EIVD |
Linke et al. 2001 |
| 14 (62±6 yr) EF=34±9% |
12 wk. Aerobic 50% VO2peak 4x45 min |
↑ VO2peak ↑ Cu/Zn SOD and GSH-Px gene expression ↔ steady-state transcript levels for eNOS, ACE, PGI2S |
Ennezat et al. 2001 |
| 10 M (55±2 yr) EF=26±3% NYHA II/III |
6 mo. Aerobic. 5x20min 70% VO2peak |
↑ VO2peak ↓ iNOS expression and protein content in skeletal muscle ↓ skeletal muscle nitrotyrosine content ↑ cytochrome coxidase activity |
Gielen et al. 2005 |
| 10 M (55±2 yr) EF=26±3% NYHA II/III |
6 mo. Aerobic. 5x20min 70% VO2peak |
↑ VO2peak ↓ TNF-α, IL-6, IL-1-β mRNA, iNOS expression in skeletal muscle ↔ serum cytokine levels |
Gielen et al. 2003 |
| 11 M (55±3 yr) EF = 21±2% NYHA II / III |
8 wk. Aerobic. 50-60% HRmax 3-5x30-60 min |
↑ 6-min walk distance ↑ forearm BF (ACh) ↔ forearm BF (NTG) ↑ L-arg transport ↔ plasma L-arginine, L-citrulline, ADMA, SDMA |
Parnell et al. 2005 |
| 12 M (55±2 yr) EF=26±3% NHYA II/III |
6 mo. Aerobic. 5x20min 70% VO2peak |
↑ VO2peak ↔ Mn and Cu/Zn SOD activity, mRNA content, or protein expression in skeletal muscle ↑ Cat activity, ↔ Cat mRNA or protein expression ↑ GPX activity, ↔ GPX mRNA or protein expression ↓ lipid peroxidation |
Linke et al. 2005 |
| 24 (55±2yr) EF: 23±1% NYHA II/III |
12 wk. Aerobic. 60-80% HRmax 5x30min |
↑ VO2peak ↓ serum TNF-α, sTNFR1, sTNFR2, IL-6. sIL-6R, sFas, sFasL |
Adamopoulos et al. 2002 |
| 23 (27-78 yr) EF: 11-45% NYHA I-IV |
4 mo. 3x/wk Aerobic+RT Aerobic: 20min 90% VT RT: 2x10 reps 50% 1RM |
↑ VO2peak ↑ NYHA class ↔ serum TNF-α, IL-6 ↓ sTNFR1, sTNFR2 (DAC) |
Conraads et al. 2002 |
| 18 (54±9 yr) NHYA 2.2±0.7 |
8 wk. Aerobic. 5x20 min 70-80% HRmax Calistenics |
↑ VO2peak ↔ plasma TNF-α, TNF-R 1 and 2, IL-6, e-selectin, sICAM, sCD14 |
Niebauer et al. 2005 |
ADMA = asymmetric dimethylarginine; CAD = coronary arterial disease; M = men; EF = ejection fraction; NYHA = New York Heart Association; EDVD = endothelium dependent vasodilation; FDVD = flow dependent vasodilation; EIVD = endothelium independent vasodilation; BF = blood flow; ACh = acetylcoline; NTG = nitroglycerine; L-NMMA = NG- monomethyl-L-arginine; SOD = superoxide dismutase; GSH-Px = glutathione-peroxidase; eNOS = endothelial nitric oxide synthase; iNOS = inducible nitric oxide synthase; ACE = angiotensin-converting enzyme; PGI2 = prostacyclin; cont = contraction; wk = week.
Although it is clear that exercise training enhances vascular endothelial function and particularly NO-mediated vasodilation, the molecular basis for this improvement has still not been completely elucidated. The preponderance of data suggests that a great part of the beneficial effects on vascular structure and reactivity are due to exercise-induced increases in endothelial shear stress. Exercise, by means of its transient increase in blood flow, affects the two forces acting on an artery. One is perpendicular to the wall and represents blood pressure, and the other acts parallel to the wall creating a frictional force at the surface of the endothelium, the so-called shear stress. Intra-arterial pressure seems to have little effect on endothelial control of vasoactivity; in contrast the principal regulator of arterial diameter is shear stress, mainly through NO release.
Acutely, shear stress mediates a biphasic production of NO with an initial rapid rise at the onset of flow, followed by a sustained increased level of production. The activation of eNOS occurs mainly through two mechanisms: calcium/ calmodulin, which dissociates eNOS from caveolin-1 [92], and phosphorylation of eNOS-S1179 and eNOS-S635 by protein kinase A, protein kinase B, and phosphoinositide-3-kinase [93, 94]. Chronically, shear stress induces an increase in the eNOS expression level and cGMP production [95]. Supporting these studies in vitro, it was demonstrated that exercise training caused an increase in eNOS expression in aortic endothelial cells [96] and prevented its decrease in the exercising muscle [97] of heart failure animal models. Recently, shear stress has also been shown to augment tetrahydrobiopterin (BH4) levels and enzymatic activity of GTP cyclohydrolase-1, the first step of BH4 biosynthesis, in human endothelial cells [98].
Although it is clear from studies in vitro and animal models in vivo that shear stress and physical exercise affect eNOS expression and BH4 levels, there is limited evidence from human studies involving CHF patients. To our knowledge, only one study has investigated this issue in humans and no changes in the steady-state transcription levels of eNOS, angiotensin converting enzyme, or prostacyclin synthase were detected in samples of vastus lateralis muscle after 12 weeks of moderate intensity endurance training [99]. On the other hand, the same study showed that exercise caused an 100% increase in gene expression of the antioxidant enzymes cupper/zinc superoxide dismutase (CuZnSOD) and glutathione peroxidise-1 (GPx-1) [99]. Thus, the positive effects of exercise on the vasculature may be due to reduced oxidative stress rather than an increase in eNOS expression, leading to an increase in NO bioavailability. However, it does not seem to be a consistent finding, since Linke et al. reported that mRNA levels and protein expression of MnSOD, CuZnSOD, GPx-1 and catalase (Cat) were unaltered after 6 months of training in samples of the vastus lateralis muscle of chronic heart failure patients [100]. These opposite findings might be due to different methods used to normalize mRNA transcripts of GPx-1 and CuZnSOD.
It is important to remember that further studies are needed to confirm these findings and also to investigate exercise induced changes in the expression and activity of eNOS and antioxidant enzymes in the endothelium and other cell types. Exercise may also affect another step involved in NO synthesis from L-arginine. After 8 weeks of endurance training at 50 to 60 % of maximum heart rate, L-arginine transport, measured by the clearance rate of L-[3H]-arginine, increased in heart failure patients associated with an improvement in endothelial function [101].
In addition to its effect on vasomotion, NO is known to antagonize key processes involved in thrombus formation, including monocyte adherence and chemotaxis, platelet adhesion and aggregation, and vascular smooth cell proliferation (Moncada et al., 2006; Brunini et al., 2007). Therefore, another potential favourable effect of exercise, though not confirmed by long-term studies, would be a reduction in the incidence of thrombotic events, which is higher in heart failure patients than in the general population. In healthy individuals, a reduction in platelet aggregation induced by oxidized LDL after 8 weeks of aerobic training was shown [102].
5.2. Exercise and Inflammation in Heart Failure
There is convincing evidence that inflammatory mediators play an important role in skeletal muscle wasting and fatigue in heart failure [103]. Among the cytokines, TNF particularly has been shown to provoke net nitrogen loss and catabolism when administered to experimental animals [104]. In skeletal muscle biopsies from heart failure patients, the cytokines interleukin IL1-β and TNF-α can induce a pathological increase in iNOS in skeletal muscle via activation of nuclear factor kappa B (NFκB) [105]. High iNOS-derived NO concentrations in skeletal muscle may inhibit key aerobic enzymes such as cytochrome c oxidase, impairing cellular respiration and causing structural damage to the mitochondria [106].
Despite all of the positive actions of NO that have been highlighted, overproduction of NO can lead to serious cytotoxic effects in the skeletal muscle and even the heart [107]. Long-term aerobic training causes a reduction in the inflammatory state of skeletal muscle, as evidenced by a diminished expression of TNF-α, IL-6 and IL-1-beta mRNA expression [108]. Consequently, there is a reduction in iNOS expression and protein nitrotyrosine content, a marker of peroxynitrite formation from NO and superoxide radical, in exercised skeletal muscle from heart failure patients [109]. These findings were inversely associated with an increase in cytocrome c oxidase activity, reflecting an improvement in muscular oxidative metabolism and increased VO2peak. Except for one paper published by Adamopoulos et al. [110], serum concentrations of cytokines and their receptors do not seem to decrease after physical training [109, 111, 112], suggesting that changes in local cytokine expression occur earlier than in the circulating system.
Plasma levels of cytokines, particularly TNF-α, IL-1 and IL-6, are elevated in CHF patients and may play a role in the genesis and progression of heart failure [113-117]. The increase in circulating concentrations of TNF-α and IL-6 correlated directly with the severity of the disease and with death and inversely with exercise tolerance in CHF [117-120]. At the cardiac level, the overexpression of cytokines, induced by a variety of stressors, promotes contractile dysfunction, impaired β adrenergic receptor sensitivity, alterations in vascular endothelium and recruitment of additional circulating inflamed cells in injured myocardium [114, 121]. Moreover, a long-term presence of these inflammatory mediators results in myocyte phenotype transition and activation of matrix metalloproteinases, which modifies the interstitial matrix, further increasing the remodeling process [122]. Studies in vivo also suggest that the death of apoptotic myocytes after sustained TNF signaling is a consequence the depletion of one or more antiapoptotic cytoprotective proteins [123].
Exercise training, particularly endurance exercise, has been shown to promote a decrease in oxidative stress and inflammation which can interfere in NOS activity and expression. Nevertheless, it is important to highlight that the benefits of exercise on vascular tone and muscle perfusion through its effects on NO bioavailability can explain only part of the benefits conferred by exercise training. Vascular tone can also be influenced by sympathetic activity and hormonal factors, and both can also be positively affected by regular exercise.
6. CONCLUSION
In this review, we critically examined the mechanisms underlying chronic heart failure and potential clinical benefits of exercise. The endothelial dysfunction present in CHF, partly reversed by exercise, is involved in widespread vasoconstriction in heart failure. It seems that interfering with L-arginine-NO pathway, inflammation and free radical metabolism may be useful adaptations to training in CHF patients.
ACKNOWLEDGEMENT
We acknowledge financial support from FAPERJ and PQ-CNPq.
REFERENCES
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]

