All published articles of this journal are available on ScienceDirect.

Primary Structure Revision and Active Site Mapping of E. Coli Isoleucyl-tRNA Synthetase by Means of Maldi Mass Spectrometry
Authors Info & Affiliations
Abstract
The correct amino acid sequence of E. coli isoleucyl-tRNA synthetase (IleRS) was established by means of peptide mapping by MALDI mass spectrometry, using a set of four endoproteases (trypsin, LysC, AspN and GluC). Thereafter, the active site of IleRS was mapped by affinity labeling with reactive analogs of the substrates. For the ATP binding site, the affinity labeling reagent was pyridoxal 5'-diphospho-5'-adenosine (ADP-PL), whereas periodate-oxidized tRNAIle, the 2',3'-dialdehyde derivative of tRNAIle was used to label the binding site for the 3'-end of tRNA on the synthetase. Incubation of either reagent with IleRS resulted in a rapid loss of both the tRNAIle aminoacylation and isoleucinedependent isotopic ATP-PPi exchange activities. The stoichiometries of IleRS labeling by ADP-PL or tRNAIleox corresponded to 1 mol of reagent incorporated per mol of enzyme. Altogether, the oxidized 3'-end of tRNAIle and the pyridoxal moiety of the ATP analog ADP-PL react with the lysyl residues 601 and 604 of the consensus sequence 601KMSKS605. Identification of the binding site for L-isoleucine or for non cognate amino acids on E. coli IleRS was achieved by qualitative comparative labeling of the synthetase with bromomethyl ketone derivatives of L-isoleucine (IBMK) or of the non-cognate amino acids valine (VBMK), phenylalanine (FBMK) and norleucine (NleBMK). Labeling of the enzyme with IBMK resulted in a complete loss of isoleucine-dependent isotopic [32P]PPi-ATP exchange activity. VBMK, NleBMK and FBMK were also capable of abolishing the activity of IleRS, FBMK being the less efficient in inactivating the synthetase. Analysis by MALDI mass spectrometry designated cysteines-462 and -718 as the target residues of the substrate analog IBMK on E. coli IleRS, whereas VBMK, NleBMK and FBMK labeled in common His-394, His-478 and Cys-718. In addition, VBMK and NleBMK, which are chemically similar to IBMK, were found covalently bound to Cys-462, and VBMK was specifically attached to His-332 (or His-337) of the synthetase. The amino acid residues labeled by the substrate analogs are mainly distributed between three regions in the primary structure of E. coli IleRS: these are segments [325-394], [451-479] and [591-604]. In the 3-D structures of IleRS from T. thermophilus and S. aureus, the [325-394] stretch is part of the editing domain, while fragments [451-479] and [591-604] representing the isoleucine binding domain and the dinucleotide (or Rossmann) fold domain, respectively, are located in the catalytic core. His-332 of E. coli IleRS, that is strictly conserved among all the available IleRS sequences is located in the editing active site of the synthetase. It is proposed that His-332 of E. coli IleRS participates directly in hydrolysis, or helps to deprotonate the hydroxyl group of threonine at the hydrolytic site.
INTRODUCTION
Aminoacyl-tRNA synthetases catalyze at the expense of ATP the activation of specific amino acids, and their subsequent transfer onto the 3'-end of homologous isoacent of L-threonine to tRNAceptor tRNAs. The specific recognition of tRNAs by aminoacyl-tRNA synthetases makes these enzymes attractive model systems for studying protein-nucleic acid interaction. To probe the structure-activity relationships of aminoacyl-tRNA synthetases, chemical modification of these enzymes with reactive analogs of the substrates has been used. Lysyl residues at the binding site for the 3'-OH acceptor arm of tRNA were identified by affinity labeling with periodate-oxidized tRNA [1-5], leading to the discovery of the KMSKS consensus sequence [3, 4, 6] characteristic of class 1 aminoacyl-tRNA synthetases. Amino acid residues at the ATP-binding site of aminoacyl-tRNA synthetases were affinity labeled with ATP derivatives such as adenosine di- or triphosphopyridoxal or 5'-p-fluorosulfonyl-benzoyladenosine [7-9]. Pyridoxal 5'-phosphate and pyridoxal 5'-diphosphate have also been used to identify the subsite for the pyrophosphate moiety or for the γ-phosphate of ATP [10, 11]. Nucleophilic amino acid residues at the binding site for L-valine or for non-cognate amino acids were identified by comparative labeling of valyl-tRNA synthetase (ValRS) with bromomethylketone derivatives [12]. The latter site referred to as the editing site is responsible for the Val/Thr editing activity of E. coli ValRS which prevents attachment of L-threonine to tRNAVal [13].
In the present work, we have applied this labeling strategy to E.coli isoleucyl-tRNA synthetase (IleRS), a monomeric synthetase with Ile/Val editing activity [14]. Reactive substrate analogs were used to identify amino acid residues at the binding site for tRNA, ATP and L-isoleucine or for non cognate amino acids on this synthetase. Lysyl residues of IleRS at the binding site for the CCA arm of tRNA have been affinity labeled by periodate-oxidized tRNAIle. In the case of the ATP-binding site, the affinity labeling reagent used was pyridoxal 5'-diphospho-5'-adenosine (or adenosine diphosphopyridoxal, ADP-PL), an adenine nucleotide analog with an adenosine diphosphate moiety and a pyridoxal moiety capable of covalently modifying amino groups in or near the phosphate binding site. The aminoacyl-tRNA synthetases such as IleRS or ValRS which are concerned with editing are believed to exhibit two catalytic sites, one for aminoacylation with the cognate amino acid, and one for editing of misactivated non-cognate amino acids. In order to identify those sites on IleRS, bromomethyl ketone derivatives of the substrate L-isoleucine (IBMK) or of non-cognate amino acids valine (VBMK), phenylalanine (FBMK) and norleucine (NleBMK), were used for qualitative comparative labeling of E. coli IleRS (Fig. 1). Bromomethyl ketone derivatives react preferentially with cysteine and histidine residues, and weakly with lysines and tyrosines. Since nucleophilic side chains are expected to be involved in the hydrolytic process at the editing site of the aminoacyl-tRNA synthetases concerned with editing, a bromomethyl moiety tethered to the carbonyl group of valine, norleucine or phenylalanine should be well situated for modification of nucleophiles at the hydrolytic site of IleRS, while the substrate analog isoleucine bromomethyl ketone would react with amino acid residues present in the binding site for L-isoleucine. The location of residues labeled by the markers is discussed on the basis of the 3-D structures of IleRS from Thermus thermophilus and Staphylococcus aureus [15, 16]. Indeed, the crystal structures at 2.5 Å resolution of IleRS from T. thermophilus in complexes with L-isoleucine and L-valine were previously reported [15], as well as that at 2.2 Å resolution of S. aureus IleRS complexed with tRNA Ile and mupirocin [16].
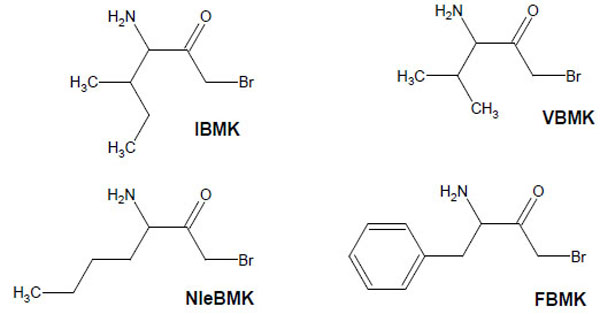
Structure of amino acid analogs used to label IleRS.
Prior to these labeling experiments, it was necessary to establish a valid primary structure of E. coli IleRS, because two different sequences had been retrieved from literature and data bases [17-19]. The correct sequence of IleRS was determined by fast peptide mapping, using a set of four different proteases to generate peptides that were identified by means of matrix assisted laser desorption-ionisation–mass spectrometry (MALDI-MS) [20].
MATERIALS AND METHODS
Materials
Isoleucyl-tRNA synthetase from E. coli strain EM20031 was purified as described [21]. The molar concentration of the synthetase was calculated from its absorbancy at 280 nm and from the following values of molecular ratio and of optical extinction coefficient deduced from amino acid composition: 105 K and 1.7 units.mg-1.cm2. tRNAIle (0.8 nmol of isoleucine acceptance/A260 unit of tRNA) was from Subriden RNA (Washington, DC). Unfractionated tRNA was obtained from the pilot facilities of the Institut de Chimie des Substances Naturelles du CNRS (Gif-sur–Yvette, France). tRNA concentration was calculated according to its specific absorption coefficient at 260 nm [22]. [14C]ATP (450 Ci/mol) was from the Commissariat à l’Energie Atomique (Saclay, France).
Sequence grade TosPheCH2Cl-treated trypsin and endoproteases LysC, GluC and AspN were from Roche Diagnostics (Meylan, France). N-(2-hydroxyethyl)piperazine-N’-2-ethanesulfonic acid (Hepes), phenylmethanesulfonyl fluoride, ribonuclease-free α-chymotrypsin, pancreatic ribonuclease (RNAse A) and diethyl pyrocarbonate (DEPC) were from Sigma. Matrix solutions and peptide standards for MALDI-MS analyses were also from Sigma. Acetonitrile (HPLC grade) was from Baker Chemical Company. All other reagents were the highest grade available.
Methods
Preparation of [14C]tRNAIle
A sample of unfractionated E. coli tRNA (16.5 mg) containing 3% of isoleucine-specific tRNA was incubated for 15 min at 25°C with 2 μM of IleRS from E. coli and 2 mM of L-isoleucine, in 5 mL of 20 mM Tris-HCl (pH 7.5) containing 2 mM ATP, 7 mM MgCl2 and 150 mM KCl. It was verified that, in these conditions, the 3% of tRNAIle present in the incubation mixture were completely charged with L-isoleucine. The sample was then incubated 30 min at room temperature, in the dark, in the presence of 10 mM NaIO4. As a consequence of the latter treatment, only the non-specific tRNAs which were not aminacylated with isoleucine by IleRS, were expected to be oxidized to a dialdehyde group on the 3'-terminal ribose. Further treatment of the mixture with 25 mM sodium borohydride provoked the reduction of the oxidized tRNAs (tRNAox) to yield tRNAox-red, tRNA molecules with primary alcohol groups (CH2OH) at the positions 2' and 3' of the ribose. Actually, these tRNAox-red species, which are not specific for isoleucine, are not concerned with further chemical modifications, because primary alcohol groups are non-reactive in the tRNA modification conditions used. The tRNA molecules specific for L-isoleucine, and which were charged with this amino acid in the presence of IleRS, were then deacylated as follows: the NaBH4-treated tRNA sample was precipitated by the addition of 0.2 volume of 5 M NaCl and 2.2 volumes of cold ethanol. After centrifugation, the pellet was dissolved in 5 mL of Tris-HCl 2 M (pH 9.0) and left 2h at 37°C. As a consequence of the latter treatment, the isoleucyl-tRNAIle species was stripped and the sample was ethanol precipitated as above. Removal of the 3'-terminal adenosine from the 3'-OH end of tRNAIle was achieved by combining periodate, lysine and alkaline phosphatase treatments, as already described [23]. The resulting tRNAIle molecules, shortened by one nucleotide at the 3'-end, were repaired with the help of tRNA nucleotidyl-transferase in the presence of [14C]ATP (9 Ci/mol) and assayed for their recovery of isoleucylation capacity, as previously described [23]. After nucleotide incorporation, the tRNA sample (including the reconstituted [14C]tRNAIle species) was separated from the excess of [14C]ATP by chromatography on a Sephadex G-50 column equilibrated with 1 mM potassium acetate buffer (pH 5.5) containing 0.1 mM MgCl2. The tRNA was then precipitated with ethanol and NaCl, as above, and stored in 1 mM potassium acetate buffer (pH 5.5) containing 0.1 mM MgCl2. The enzymatic labeling of truncated tRNAIle at its 3'-end, followed by the chromatographic step, yielded 13.7 mg of total tRNA containing 3% of [14C]tRNAIle species, which corresponded to a yield of 83% of the initial amount of unfractionated E. coli tRNA (16.5 mg). The specific radioactivity of [14C]tRNAIle molecules in the mixture, as measured by trichloroacetic acid precipitation, was 9,000 counts per minute per nanomole of radiolabeled tRNA.
Inactivation and Labeling of IleRS by Periodate-Oxidized tRNAIle
tRNAIle (0.8 nmol of isoleucine acceptance/A260 unit of tRNA) or [14C]tRNAIle (specific radioactivity 9,000 counts.min-1.nmole-1) were oxidized with NaIO4 as in [1]. Kinetics of inactivation of IleRS (1 μM) by periodate-oxidized tRNAIle (tRNAIleox 4 μM) were followed at 37°C in 20 mM imidazole hydrochloride (pH 8.0) containing 25% glycerol, 10 mM MgCl2, and 2 mM NaBH3CN. At various times, 10 μL aliquots of the incubation mixture were diluted in imidazole hydrochloride buffer (pH 7.6) containing 10 mM 2-mercaptoethanol, 0.1 mM EDTA and 200 μg/mL bovine serum albumin, and assayed for isoleucine-dependent isotopic ATP-PPi exchange and tRNAIle aminoacylation activities [24, 25].
For stoichiometry measurements, IleRS (3 μM) was incubated with 396 μM of the unfractionated tRNA sample containing 12 μM of periodate-oxidized [14C]tRNAIle ([14C]tRNAIleox) (specific radioactivity 9,000 counts.min-1.nmole-1), in the same conditions as above. At various times, portions of the incubation mixture were withdrawn and assayed for enzymatic activity. In parallel, aliquots were submitted to ribonuclease A digestion (10 μg/mL) for 2 min. Incorporation within the enzyme of the [14C]radioactivity was followed by precipitation with 5% trichloroacetic acid, as already described [1].
Preparation of IleRS-[14C]tRNAIleox Covalent Complex
IleRS (3 μM in 5 mL) was incubated with 12 μM periodate-oxidized [14C]tRNAIle (specific radioactivity 9,000 counts.min-1.nmole-1) in the same conditions as above, and the [14C]adenosine incorporation in the enzyme was followed by submitting aliquots of the incubation mixture to ribonuclease A digestion and trichloroacetic acid precipitation [1]. At 150 min, when 97% of the initial IleRS activity was lost, while 1 mol of [14C]tRNAIleox was incorporated per mol of enzyme, the reaction was quenched by the addition of 25 mM NaBH4. In order to separate the IleRS-[14C]tRNAIleox covalent complex from the excess of tRNA, the reaction mixture was loaded onto a Sephadex G-100 column equilibrated with 0.1M ammonium acetate (pH 8.0). Elution was monitored by radioactivity measurement. The IleRS-[14C]tRNAIleox complex was vacuum-dried.
Preparation and Purification of [14C]tRNAIleox-Labeled Peptides of IleRS
The labeled IleRS was digested with α-chymotrypsin for 4h at 37°C, at a protease to synthetase ratio of approximately 1/50 (w/w). The chymotryptic digestion was stopped by adding phenylmethanesulfonyl fluoride (PMSF) at a final concentration of 0.5 mM. Peptide purification from the chymotryptic digest of the IleRS-[14C]tRNAIleox covalent complex was performed by a two-step chromatography on Sephadex G-50 as already described [3-5], followed by further purification by reverse-phase high-performance liquid chromatography (RPLC) on a Merck Superspher RP18 column (250 x 4 mm, 4 μm particles, 10 nm pore size). Peptides were eluted with a linear gradient (from 0 to 80% B) with the following eluents: A, 0.1% trifluoroacetic acid in water; B, 0.1% trifluoroacetic acid and 80% acetonitrile in water.
Inactivation and Labeling of IleRS by Pyridoxal 5'-diphospho-5'-adenosine
Pyridoxal 5'-diphospho-5'-adenosine (ADP-PL) was synthesized and purified as already described [26]. Its molar absorption coefficients in 0.1 N NaOH were 5060 M-1.cm-1 at 394 nm and 5550 M-1.cm-1 at 393 nm, respectively. Inactivation of IleRS (6 μM) by ADP-PL (1 mM) was carried out at 37°C, in 50 μL of 50 mM Hepes/Na (pH 7.8). At various times, 5-μL portions of the incubation mixtures were withdrawn and quenched by the addition of 10 μL of freshly prepared 0.1 M NaBH4 in 50 mM Hepes/Na (pH 7.8), followed by dilution with 0.5-1 mL Tris/HCl pH 7.6, containing 10 mM 2-mercaptoethanol, 0.1 mM EDTA, and 200 μg/mL bovine serum albumin. The diluted portions were assayed for the isoleucine-dependent isotopic ATP-PPi exchange and tRNAIle aminoacylation activities [24, 25].
Incorporation of ADP-PL into IleRS was monitored as already described [7]. IleRS (6 μM) was incubated at 37°C in 50 μL of 50 mM Hepes/Na (pH 7.8) containing 0.2-3 mM ADP-PL. After 90 min, each incubation mixture was added to 10 μL of freshly prepared 0.1 M NaBH4 in 50 mM Hepes/Na (pH 7.8). 5 μL of each NaBH4-treated sample was diluted and assayed for the isoleucine-dependent isotopic ATP-PPi exchange and tRNAIle aminoacylation activities [24, 25]. In parallel, a 50 μL aliquot was analysed by size exclusion chromatography on a TSK 4000 PW column (300 mm x 7.5 mm) with 0.2 M ammonium acetate (pH 7.5) as eluent, to separate the covalently bound and the unbound NaBH4-reduced ADP-PL. Elution was monitored by absorbance measurement at 325 nm; a molar absorption coefficient of 8800 M-1.cm-1 (pH 7.5) at 325 nm was used to quantify the amount of pyridoxyl group covalently bound to the enzyme [26-28].
Inactivation of IleRS by Bromomethyl Ketone Derivatives of Amino Acids
Bromomethyl ketone derivatives of amino acids were synthesized as already described [12]. Isoleucyl-tRNA synthetase (IleRS), stored in 20 mM Tris-HCl buffer (pH 7.8) containing 50% glycerol and 10 mM 2-mercaptoethanol, was extensively dialyzed against the appropriate buffer prior to incubation with a given reagent. Inactivation of IleRS (2 μM) was carried out in the presence of either of the bromomethyl ketone derivatives (Fig. 1) of various amino acids (IBMK, VBMK, NleBMK or FBMK) at a final concentration of 2 mM, at 37°C in 50 μl of 50 mM Hepes/Na, pH 7.8.
At various times, 5 μL aliquots of the incubation mixtures were withdrawn and quenched by a 100 fold dilution with 20 mM Tris-HCl buffer (pH 7.8) containing 200 μg/mL of bovine serum albumin and 50 mM 2-mercaptoethanol. The diluted portions were assayed for the isoleucine-dependent isotopic ATP-PPi exchange and tRNAIle aminoacylation activities [24, 25].
Inactivation of IleRS by Diethyl Pyrocarbonate
Inactivation of IleRS (2 μM) with 0.5 mM diethyl pyrocarbonate (DEPC) was performed in 50 mM potassium phosphate buffer (pH 6.5) by adding the reagent dissolved in absolute ethanol to the enzyme solution. Final ethanol concentration was less than 2%, which had no effect on enzyme activity. The controls contained equal volumes of ethanol. The concentration of DEPC solutions was deduced by reaction with free imidazole of known concentration, using a molar absorption coefficient at 240 nm and pH 6.5 of 3200 M-1.cm-1. At various times, 5 μL aliquots of the incubation mixture were withdrawn and diluted at least 100 fold with 20 mM Tris-HCl buffer (pH 7.8) containing 200 μg/mL of bovine serum albumin and 10 mM 2-mercaptoethanol.
Proteolytic Digestion and Peptide Separation
After 1-2 hours of incubation of IleRS with the bromomethyl ketone derivatives of amino acids, or with DEPC, when the assayed diluted portions had reached the maximum inactivation level, the incubation mixtures were extensively dialyzed against 0.1 M ammonium acetate (pH 8.0). Whatever the used reagent, labeled IleRS was digested overnight at 37°C with TosPheCH2Cl-treated trypsin at a protease to substrate ratio of 1/20 (w/w).
ADP-PL-labeled peptides were purified at room temperature on a Vydac C18 reverse-phase column (150 mm x 2.1 mm; 5 μm particles), at a flow rate of 0.25 mL/min (Applied Biosystems model 140A HPLC gradient system, and model 1000S diode array detector). Peptides were eluted with a linear gradient composed of the following solutions: solution A, 0.1% TFA in water; solution B, 0.1% TFA and 80% acetonitrile in water and 0.25 mL fractions were collected.
Peptides labeled with IBMK, VBMK, NleBMK, FBMK were separated on a BioCad Sprint (Perkin Elmer Biosystems) HPLC system equipped with a C18 reverse-phase column (4.6 x 250 mm, 5 μm particles from Interchim), at a flow rate of 1 mL/min. Tryptic digests were injected into the column equilibrated with 0.1% TFA in water (solvent A). Linear gradients of acetonitrile were developed from 10% to 100% B during 60 min (solvent B: 10% solvent A in acetonitrile) and 1 mL fractions were collected.
In the case of DEPC-labeled peptides, in order to prevent the removal of ethoxyformyl groups during the chromatographic separation, elution was achieved with linear gradients of acetonitrile in 1mM potassium phosphate buffer (pH 7.0). As a control, the ethoxyformyl groups of DEPC-labeled peptides was removed by hydroxylamine as follows: vacuum-dried DEPC-labeled peptides were dissolved in 0.1 M Tris-HCl (pH 7.0) containing 0.1 M hydroxylamine adjusted to pH 7.0. Under this condition, optimal removal of the ethoxyformyl group was obtained after 30-60 min of incubation at 28°C.
Mass Spectrometry
Peptide samples were prepared by mixing 1 μL of matrix solution [100 mM α-cyano-4-hydroxy-cinnamic acid (HP G2037A or Sigma) in 50 % acetonitrile, 0.1% trifluoroacetic acid] with 1 μL peptide solution (1-10 pmol). This mixture was then loaded on the probe tip and dried.
MALDI mass spectra of labelled peptides were obtained in the linear mode with time of flight mass spectrometers (Hewlett Packard G2025 or MicromassVG Tofspec). From 100 to 200 shots were accumulated for each spectrum acquisition in the positive-ion mode. Calibration with external standards was obtained with a mixture of peptide standards (HP G2052A) in the range 1000-7000 Da. High resolution MALDI-MS of IleRS digests was performed with a Bruker Reflex III mass spectrometer operated in the reflectron mode with the same matrix as above. For sequence confirmation, post source decay (PSD) spectra of fragment ions from a selected precursor were acquired in 11-13 voltage steps, starting at 30 kV reflector voltage, with an ion gate operated at a resolution of approximately 150.
N-Terminal Sequencing
A Model 470A sequencer (Applied Biosystems) was used for automated Edman degradation of purified [14C]tRNAox-labeled peptides. Samples (30 μL aliquots, 100-200 pmol) were loaded on glass fiber filters that had been previously treated with 2.5 mg of Biobrene (Applied Biosystems) and submitted to three precycles. The 03RPTH program, modified for our on-line PTH identification setup, was used for sequencing. Automated on-line PTH analysis was performed as described in [29].
RESULTS
Correction of E. coli IleRS Sequence
Two IleRS sequences were retrieved from databases. The first sequence was published as an aminoacid sequence only [17], whereas the second sequence [18], is a nucleotidic sequence. As revealed by protein sequence alignment, there are 15 stretches where conflicts occur between these sequences. A previous MALDI-MS peptide mapping experiment demonstrated that none of these primary structures is correct. These findings were reinforced by codon usage analysis and multiple alignment with IleRS sequences from other species [30].
A correct sequence was then definitely established by means of peptide mapping using a set of four endoproteases (trypsin, LysC, AspN and GluC) and MALDI-MS analysis of the released peptides. When necessary, sequence confirmation was obtained by means of PSD fragment analysis [20]. Thus, the first sequence [17] had 6 errors, while the second [18] had 9 wrong sequence stretches. The correct sequence (937 residues, average molecular mass 104362 Da) is given in Fig. 2, and primary structure numbering given in this work refers to this sequence only.

Inactivation and Labeling of IleRS by Periodate-Oxidized [14C]tRNAIle
The isoleucine-specific component of unfractionated E. coli tRNA representing 3% of the total tRNA, was charged with L-isoleucine, followed by periodate oxidation of the non-specific tRNAs which were not aminacylated with isoleucine, to yield tRNAox. The tRNAox molecules were further reduced to tRNAox-red with sodium borohydride, while the tRNA molecules specific for L-isoleucine, and which were charged with this amino acid in the presence of IleRS, were deacylated and converted to [14C]tRNAIle by combining periodate, lysine, alkaline phosphatase and nucleotidyl-transferase treatments, as already described [23]. Altogether, these treatments yielded [14C]tRNAIleox molecules with a specific radioactivity of 9,000 counts.min-1.nmole-1, in a mixture with various tRNAox-red species.
When IleRS (1 μM) was incubated with 4 μM of periodate-oxidized tRNAIle (tRNAIleox), the isoleucine-dependent isotopic [32P]PPi-ATP exchange and tRNAIle aminoacylation activities of the synthetase were lost by more than 95% (Fig. 3). The kinetics of inactivation fitted single exponential curves, with an estimated rate constant of inactivation of 0.037 min-1 and 0.025 min-1 in the tRNAIle aminoacylation and isoleucine-dependent isotopic [32P]PPi-ATP exchange activities, respectively (Fig. 3). Intact E. coli tRNAIle at a final concentration of 10 μM fully protected the enzyme (1 μM) against inactivation and labeling by tRNAIleox (4 μM). Combination of L-isoleucine (8 mM) and ATP-Mg2+ (8 mM) also afforded full protection (Fig. 3).
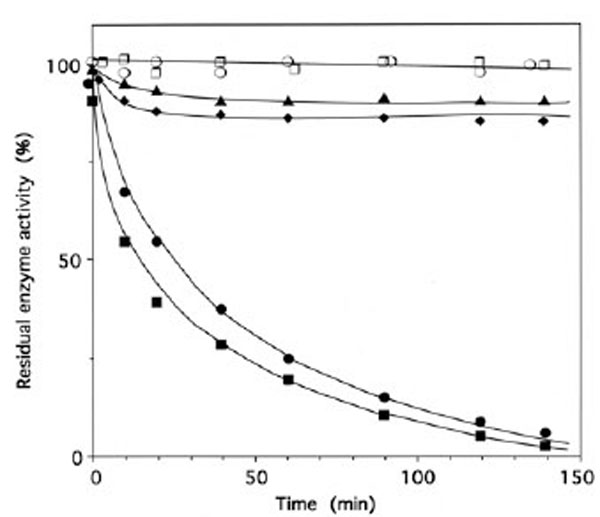
Time course of inactivation of IleRS by tRNAIleox. IleRS (1 μM) was incubated with 4 μM tRNAIleox, as described under Methods. The residual isoleucine-dependent isotopic [32P]PPi-ATP exchange (●, ▲ ○) and tRNAIle aminoacylation (■ ◆, ◻) activities were measured as a function of time. The control experiments (○ , □) were without tRNAIleox or with 4 µM intact tRNAIle instead of tRNAIleox. For the protection experiments (▲ ◆), 20 µM of intact tRNAIle was present with the enzyme prior to tRNAIleox addition. In another protection experiment, IleRS (1 µM) was pre-incubated with a combination of 8 mM MgATP and 8 mM L-isoleucine, prior to tRNAIleox addition. In the latter case, kinetics of enzyme activity loss for isoleucine-dependent isotopic [32P]PPi-ATP exchange and tRNAIle aminoacylation activities were superimposable on those of the controls (○ , □).
Similarly, when IleRS (3 μM) was incubated in the presence of 396 μM of unfractionated tRNAox-red sample containing 12 μM of periodate-oxidized [14C]tRNAIle, the isoleucine-dependent isotopic [32P]PPi-ATP exchange and tRNAIle aminoacylation activities of the synthetase were lost, in parallel with the incorporation of (1.10 ± 0.05) mol of [14C]tRNAIleox per mol of E. coli IleRS (Fig. 4), in agreement with the 1:1 enzyme:tRNA active stoichiometry. Treatment with NaBH4 (5 mM) of the tRNA sample containing the [14C]tRNAIleox species, prior to incubation with IleRS, prevented inactivation and labeling of the synthetase, suggesting that only the periodate-oxidized isoleucine-specific component of the total tRNA specifically reacts with this synthetase.
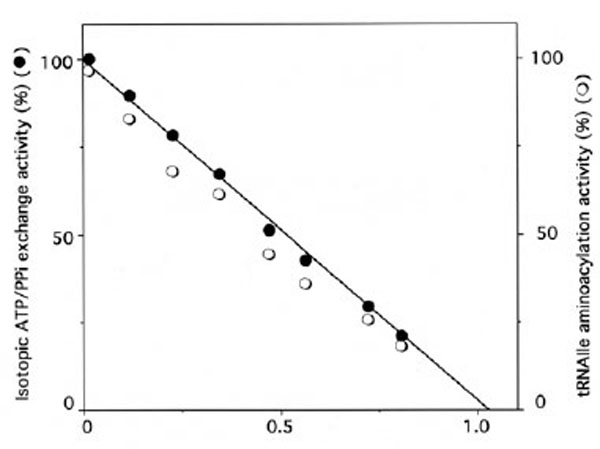
Stoichiometry of labeling of IleRS by [14C]tRNAIleox.IleRS (3 µM) was incubated with 396 μM of unfractionated tRNA sample containing 12 µM of [14C]tRNAIleox (specific radioactivity 9,000 counts.min-1.nmole-1). The number of [14C]tRNAIleox molecules (n) incorporated per mol of enzyme is represented as a function of the residual isoleucine-dependent isotopic [32P]PPi-ATP exchange (●) and tRNAIle aminoacylation (○) activities. The control experiments in the presence of intact [14C]tRNAIle showed no incorporation of radioactivity.
Altogether, these results strongly suggest that the oxidized CCA-arm of enzyme-bound tRNAIle actually reacts with lysyl residues at or near the catalytic center of E. coli IleRS.
Inactivation and Labeling of IleRS by Pyridoxal 5'-diphospho-5'-adenosine
Upon incubation with 1 mM ADP-PL, followed by reduction with sodium borohydride, the isoleucine-dependent isotopic [32P]PPi-ATP exchange and tRNAIle aminoacylation activities of IleRS (6 μM in 50 μL) were lost by more than 97% within 30 min (Fig. 5), in parallel with the incorporation of (0.98 ± 0.03) mole of ADP-PL/mole of IleRS (Fig. 6).
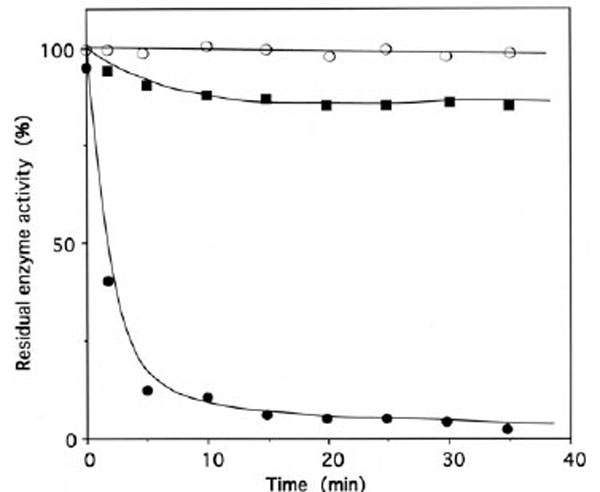
Time course of inactivation of IleRS by ADP-PL.
IleRS (6 µM) was incubated with 1 mM ADP-PL, as described under Methods. Kinetics of enzyme activity loss were superimposable for isoleucine-dependent isotopic [32P]PPi-ATP exchange and tRNAIle aminoacylation activities (● , ○ , ■). The control experiments (○) were without ADP-PL or with 1 mM ATP instead of ADP-PL. For the protection experiments (■), the enzyme was preincubated with 8 mM L-isoleucine and 8 mM ATP-Mg2+ prior to ADP-PL addition.
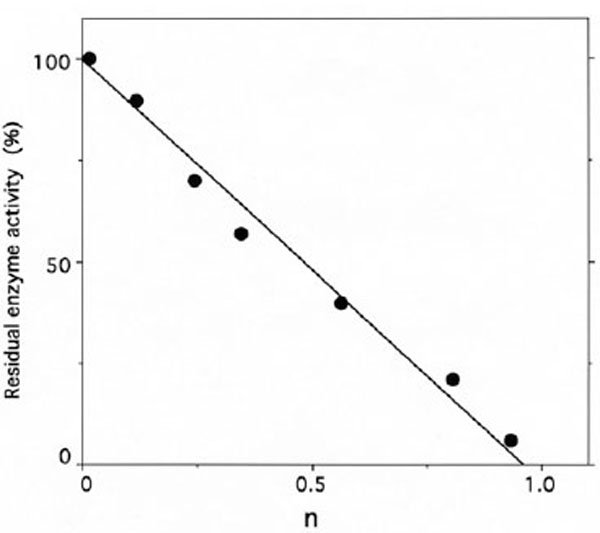
Stoichiometry of labeling of IleRS by ADP-PL.
IleRS (6 µM) was incubated with 0.2-3 mM ADP-PL, as described under Methods. The residual enzyme activity is plotted against the molar fraction of ADP-PL incorporated per mole of enzyme (n), as deduced from the absorbance at 325 nm of NaBH4-reduced ADPPL covalently bound to IleRS and separated from the unbound reagent by chromatography through a TSK 4000 PW column.
These results agree with the known presence of one binding site for MgATP on IleRS. The kinetics of inactivation fitted a single exponential curve, with an estimated rate constant of inactivation of the tRNAIle aminoacylation and isoleucine-dependent isotopic [32P]PPi-ATP exchange activities of 0.4 min-1 (Fig. 5). Combination of L-isoleucine (8 mM) and ATP-Mg2+ (8 mM) protected the enzyme against inactivation by ADP-PL (Fig. 5).
As shown in Fig. (7), the relationship between the fraction of inactivated IleRS and the concentrations of free ADP-PL in the incubation mixtures is hyperbolic, indicating that a reversible enzyme-reagent complex is formed prior to the labeling. From the saturation curve obtained, the dissociation constant for the non-covalent enzyme-reagent intermediate was estimated to be 0.17 mM, which is similar to the Kd value (0.176 mM) of ATP binding to IleRS [31]. Altogether, these results suggest that, when bound to IleRS, ADP-PL is well fitted to the ATP-binding site.
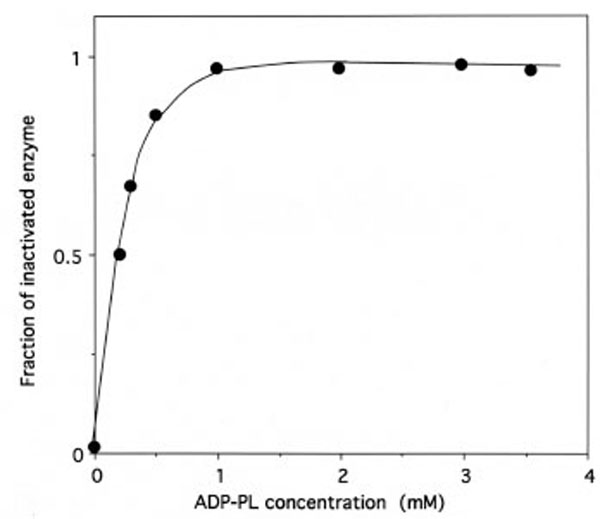
Saturation of IleRS by ADP-PL.
The incubation mixtures were the same as in Figure 5. The fraction of inactivated IleRS, as deduced from kinetics data from Figure 5, is represented as a function of free ADP-PL concentration. From this saturation curve, the half-effect concentration of ADP-PL in the inactivation of IleRS was estimated to be 0.17 mM.
Inactivation of IleRS with Bromomethyl Ketone Derivatives of Various Amino Acids
More than 97% of the isoleucine-dependent isotopic [32P]PPi-ATP exchange activity of E.coli IleRS (2 μM in 50 μL) was lost within 30 min, upon incubation with 2 mM IBMK, with an estimated rate constant of inactivation of 0.34 min-1 (Fig. 8). The effects of substrates on the inactivation of IleRS by IBMK were examined (Fig. 8). The presence of 8 mM MgATP provided substantial protection, while the presence of 8 mM L-isoleucine alone or in combination with 8 mM MgATP afforded full protection. Protection of IleRS by L-isoleucine against inactivation by IBMK was not the result of the reaction of this reagent with the α-amino group of the amino acid, since 8 mM L-valine afforded no protection (Fig. 8).
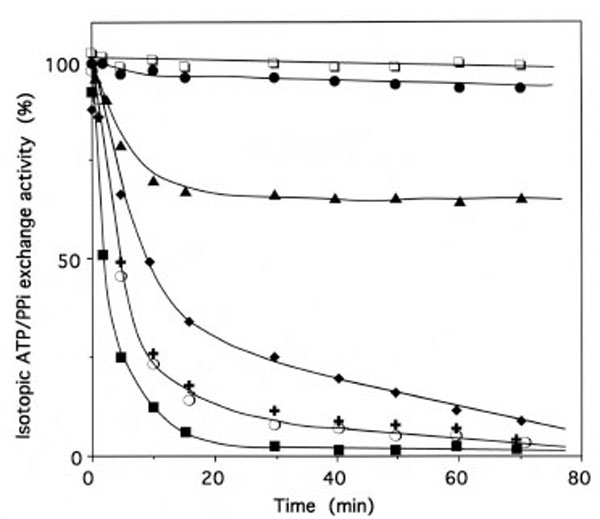
Inactivation of IleRS by bromomethyl ketones derived from amino acids.
The enzyme (2 µM) was incubated at 37°C in 50 mM Hepes/Na (pH 7.8), with IBMK (■), NleBMK (+), VBMK ○ or FBMK (◆), at a final concentration of 2 mM. Control experiments (□) were carried out in the absence of reagent. Aliquots of the incubation mixtures were withdrawn, quenched by dilution with a buffer that contained 50 mM 2-mercaptoethanol, and assayed for the isoleucine-dependent isotopic [32P]PPi-ATP exchange activity, as described under Methods. For the protection experiments, the enzyme was pre-incubated with 8 mM MgATP (▴) or 8 mM Lisoleucine (●), prior to IBMK addition. The combination of 8 mM MgATP and 8 mM L-isoleucine was as efficient as 8 mM Lisoleucine alone in the protection of IleRS against inactivation by IBMK. Pre-incubation of IleRS with 8 mM L-valine prior to IBMK addition did not affect the kinetics of inactivation of the synthetase by this cognate amino acid analog (■).
Data are plotted as the percent of activity remaining versus incubation time.
The relationship between the fraction of IBMK-inactivated IleRS and the concentrations of IBMK in the incubation mixtures is hyperbolic (Fig. 9), indicating that a reversible IleRS/IBMK complex is formed prior to the labeling. From the saturation curve obtained, the half-effect concentration of IBMK in the inactivation of IleRS was estimated to be 0.1 mM. Even though the latter value is much larger than the Kd value (6.8 μM) of isoleucine binding to IleRS [31], this saturation behavior might reflect that, when bound to IleRS, IBMK is well fitted to the isoleucine-binding site. Altogether, these results would indicate that the substrate analog IBMK reacts with nucleophilic amino acid residues at or near the isoleucine binding site of IleRS.
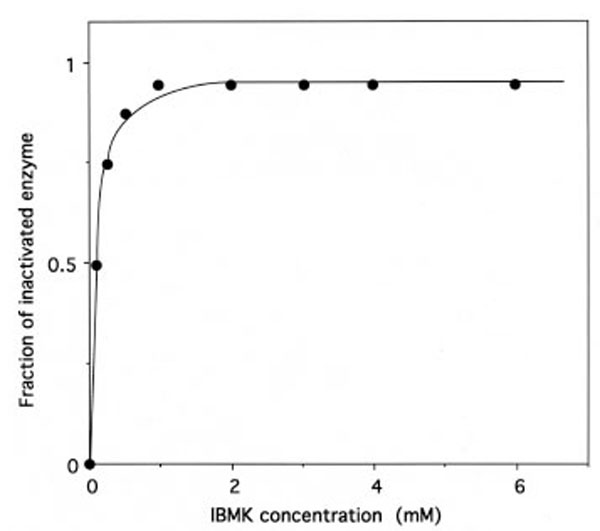
Saturation of IleRS by IBMK.
The incubation mixtures were the same as in Figure 8. The fraction of IBMK-inactivated IleRS, as deduced from kinetics data from Figure 8, is represented as a function of free IBMK concentration. From this saturation curve, the half-effect concentration of IBMK in the inactivation of IleRS was estimated to be 0.1 mM.
Similarly, incubation of IleRS with either of the used bromomethyl ketone derivatives of non-cognate amino acids resulted in a time-dependent inactivation of the isoleucine-dependent isotopic [32P]PPi-ATP exchange activity. However, under the same conditions, the estimated rate constants of inactivation of IleRS (2 μM) by the latter reagents at a final concentration of 2 mM were lower than that of IBMK (0.15, 0.15, and 0.072 min-1 for VBMK, NleBMK and FBMK, respectively), versus 0.34 min-1 for (IBMK) (Fig. 8), suggesting that these reagents are not equally accessible to the target residues in the active site of the enzyme.
As a control, the effect on IleRS activity of diethylpyrocarbonate (DEPC), a reagent having a structure unrelated to amino acids, which reacts preferentially with histidine side chains, was also examined. Incubation of IleRS (2 μM in 50 μL) with 0.5 mM DEPC resulted in a complete inactivation of the synthetase (results not shown). L-isoleucine (2 mM) or L-isoleucine (2 mM) associated to MgATP (2 mM) afforded full protection against inactivation of IleRS by DEPC (results not shown).
Altogether, these results suggest that nucleophilic amino acid residues critical for activity such as cysteines or histidines, located within the enzyme active site or in the vicinity thereof are the targets of the various used reagents.
Preparation of Labeled Peptides of IleRS
A covalent IleRS-[14C]tRNAIleox complex (96% inactivated) was digested with ribonuclease-free chymotrypsin. In order to counterselect the tRNA-labeled chymotryptic peptides from the unlabeled ones, the digest was fractionated in two steps by size exclusion chromatography on Sephadex G-50, as already described [3-5]. In the first step, the [14C]tRNA-labeled peptides in which the tRNA moiety had remained intact eluted as a single peak in the void volume of the first Sephadex G-50 column, as expected. This step separated the tRNA-bound peptides from the bulk of smaller unlabeled ones. In the second step, the radioactive material recovered from the first step was treated with ribonuclease A and loaded on a second Sephadex G-50 column. This second chromatography separated the [14C]adenosine-labeled peptides from the large unlabeled ones, which might have coeluted in the void volume of the first column [3-5]. [14C]Adenosine-labeled peptides eluted as a single peak which was recovered for further purification by reverse-phase high-performance liquid chromatography (RPLC) on a Superspher RP18 column. The majority of the labeled material (85% of the applied radioactivity) was recovered as a single peak close to the void volume of the RPLC column (results not shown).
Other labeled peptides were obtained from IleRS samples that have been inactivated by 96%, 98%, 98%, 97%, 90% and 97%, in the presence of ADP-PL, IBMK, VBMK, NleBMK, FBMK and DEPC, respectively. Tryptic digests of labeled IleRS were separated by RPLC. As a control, an intact IleRS sample was submitted to trypsin digestion and RPLC separation in the same conditions as the labeled synthetase.
Peptides (215 nm absorbing fractions) obtained in a labeling experiment were analysed by MALDI-MS. Similarly, the corresponding 215 nm absorbing fractions of the entire RPLC chromatographic profile of the control were also analysed by MALDI-MS. Distinction between labeled and unlabeled peptides, as well as assignment of the ADP-PL-, IBMK-, VBMK-, NleBMK-, FBMK- or DEPC-labeled peptides were achieved by comparing the molecular masses of fractions containing 215 nm absorbing material obtained in a labeling experiment to those of the corresponding fractions in the control. In the cases of ADP-PL- or FBMK- labeled peptides, peptide elution was also monitored at 260 nm (adenine or Phe detection).
Analysis of Labeled Peptides by MALDI-MS or Automated Edman Degradation
Labeled peptides were identified by means of MALDI-MS analysis, using the mass increase values corresponding to the binding of ADP-PL, IBMK, NleBMK, FBMK, VBMK and DEPC (561.1, 127.1, 127.1, 161.1, 113.1 and 72.1 Daltons, respectively; cf Fig. 1). Mass values of IleRS labeled peptides are given in Table 1. Accordingly, ADP-PL-labeled peptides corresponded respectively to stretches 601KMSK604 and 602MSKSIGNTVSPQDVMNK618 in the primary structure of E. coli IleRS (Fig. 2). Thus, the two lysyl residues of the consensus sequence 601KMSKS605 were labeled with this reactive ATP analog.
Matrix Assisted Laser Desorption-Ionization Mass Spectrometry (MALDI-MS) of ADP-PL-, IBMK-, VBMK-, NleBMK-, FBMK- and DEPC-Labeled Peptides of IleRS
| Labeled Peptide Sequence | Measured Mass | Calculated Mass | Mass Diff. | Labeled Residue |
|---|---|---|---|---|
| ADP-PL-labeled peptides | ||||
| 601KMSK604 | 1055.1 | 493.6 | 561.5 | K601 |
| 602MSKSIGNTVSPQDVMNK618 | 2398.7 | 1837.1 | 561.6 | K604 |
| IBMK-labeled peptides | ||||
| 451IESMVANRPDWCISR465 | 1904.9 | 1777.1 | 127.8 | C462 |
| 717FCSVEMVSFYLDIIK731 | 1921.5 | 1794.2 | 127.3 | C718 |
| VBMK-labeled peptides | ||||
| 305FTHPFMG......DYVIGQK34 | 4574.5 | 4461.0 | 113.5 | ? |
| 325DAGTGA…PGHGPDDY342 | 1851.7 | 1738.7 | 113.0 | H332/337 |
| 390GALLHVEK397 | 980.5 | 867.0 | 113.5 | H394 |
| 451IESMVANRPDWCISR465 | 1890.6 | 1777.1 | 113.5 | C462 |
| 468TWGVPMSLFVHK479 | 1515.5 | 1402.7 | 112.8 | H478 |
| 717FCSVEMVSFYLDIIK731 | 1907.7 | 1794.2 | 113.5 | C718 |
| NleBMK-labeled peptides | ||||
| 390GALLHVEK397 | 993.6 | 867.0 | 126.6 | H394 |
| 451IESMVANRPDWCISR465 | 1904.7 | 1777.1 | 127.6 | C462 |
| 468TWGVPMSLFVHK479 | 1529.1 | 1402.7 | 126.4 | H478 |
| 717FCSVEMVSFYLDIIK731 | 1921.8 | 1794.2 | 127.6 | C718 |
| FBMK-labeled peptides | ||||
| 390GALLHVEK397 | 1027.7 | 867.0 | 160.7 | H394 |
| 468TWGVPMSLFVHK479 | 1563.5 | 1402.7 | 160.8 | H478 |
| 717FCSVEMVSFYLDIIK731 | 1955.1 | 1794.2 | 160.9 | C718 |
| DEPC-labeled peptides | ||||
| 583APYRQVLTHG…KMSK604 | 2550.0 | 2477.8 | 72.2 | H591 |
| 890VALS…CWHYTQDVGK912 | 2677.3 | 2605.0 | 72.3 | H905 |
Masses, corresponding to [M + H]+ ions, are calculated (unlabeled peptides), or observed. Mass differences correspond to the covalent addition of one reagent (theoretical values are 561.1 for ADP-PL, 127.1 for L-isoleucine, 113.1 for L-valine, 127.1 for L-norleucine, 161.1 for L-phenylalanine and 72.0 Da for DEPC, cf Fig. 1).
In total, peptides labeled by bromomethyl ketone derivatives of amino acids correspond respectively to stretches 305FTHPFMG....DYVIGQK347, 390GALLHVEK397, 451IESM VANRPDWCISR465, 468TWGVPMSLFVHK479 and 717FCS VEMVSFYLDIIK731 in the primary structure of E. coli IleRS. Among these, VBMK-labeled fragment 305-347 contains four histidyl residues of which only one was expected to be attached to the label (Table 1). In order to identify the labeled histidyl residue of this fragment, this VBMK-labeled peptide was further digested with chymotrypsin, and analysed by MALDI-MS. Only one modified peptide was found at m/z 1851.7 Da, corresponding to the mass of fragment 325-342 (calculated mass 1738.7) modified with one valyl moiety (mass adduct 113.1). As a consequence, the labeled histidyl residue of the parent VBMK-labeled fragment 305-347 is His-332 or His-337.
Thus, the amino acid residues of E. coli IleRS labeled by the amino acid-derived bromomethyl ketones were His-332 (or His-337), His-394, Cys-462, His-478 and Cys-718.
DEPC-labeled peptides corresponded respectively to positions 583APYRQVLTHGFTVDGQGRKMSK604 and 890VALSKAEGEKCPRCWHYTQDVGK912 in the primary structure of E. coli IleRS (Table 1), and the labeled residues were thus His-591 and His-905, respectively. Identification of the labeled histidine residues was ascertained by removal of the ethoxyformyl groups of DEPC-labeled peptides with the help of 0.1 M hydroxylamine in 0.1 M Tris-HCl (pH 7.0), as described under Methods, followed by MALDI-MS analysis of the recovered unlabeled peptides.
When the [14C]tRNAIleox-labeled peptides of IleRS, eluted as a single peak close to the void volume of the C18 RPLC column, were analyzed by MALDI mass spectrometry, no signal could be found. Therefore, the peptidic material of this peak was submitted to N-terminal sequencing by automated Edman degradation. A unique [14C]tRNAox-labeled peptide was found in this peak, the sequence of which was 603SKSIGN608. Thus, the tRNAIleox-labeled residue of IleRS was Lys-604.
DISCUSSION
Primary Structure Revision of E. coli IleRS
Most of the errors in the two published IleRS primary structures are related to shifts in the nucleotide reading frame. The two wrong sequences were left as such for a long time, probably because the first sequence was published as a protein sequence only [17] and the second sequence [18] was not cross-checked with the first one. The combination of peptide mass mapping by MALDI-MS and PSD fragmentation of peptide ions was a fast way to determine the correct sequence. The involvement of a set of four different proteases was required for a total sequence coverage of this 104 kDa enzyme. Cleavage specificities of these proteases were helpful for solving single residue conflicts such as C vs R at location 586, Q vs E at location 636 and N vs K at location 829.
Analogy with the 3-D Structures of IleRS from Thermus Thermophilus and Staphylococcus aureus
The crystal structures at 2.5 Å resolution of IleRS from T. thermophilus in complexes with L-isoleucine and L-valine were previously reported [15], as well as that at 2.2 Å resolution of S. aureus IleRS complexed with tRNAIle and mupirocin [16].
Alignment of the amino acid sequence of several IleRS from different origins reveals an average of 35 % identities or conservative replacements (results not shown). Since it is generally believed that proteins sharing more than 30% primary structure similarity are somewhat similarly folded, it can be reasonably expected that the isoleucyl-tRNA synthetases from E. coli, T. thermophilus and S. aureus present similar overall three-dimensional structures. Therefore, our labeling data can be discussed on the basis of the 3-D structures of IleRS from T. thermophilus and S. aureus [15, 16].
The crystal structure of S. aureus IleRS is composed of three regions of differing functions: a region that binds the tRNA anticodon loop, a catalytic core that activates and transfers the amino acid and an editing domain that removes any incorrectly incorporated amino acid [16]. The amino acid residues labeled by tRNAIleox or ADP-PL, as well as by IBMK, NleBMK, FBMK or VBMK, or the histidine-specific reagent DEPC, are mainly distributed between four regions in the primary structure of IleRS: these are fragments [325-394], [451-479], [591-604] and [718-905] (Table 1). In the 3-D structures of IleRS from T. thermophilus and S. aureus, fragment [325-394] is part of the editing domain, while fragments [451-479] and [591-604] representing the isoleucine binding domain and the dinucleotide (or Rossmann) fold domain, respectively, are located in the catalytic core.
All the thiol-specific reagents used (including the substrate analog IBMK) were found covalently attached to Cys-718 (Table 1), in the C-terminal region of E. coli IleRS. These results would suggest that Cys-718 was unspecifically labeled, due to its special reactivity toward alkylation by the bromoketone moiety of the amino acid analogs.
The Binding Site for ATP or for the 3'-end of tRNA on E. coli IleRS
Several observations suggest that ADP-PL and tRNAIleox, are affinity labeling reagents for E. coli isoleucyl-tRNA synthetase. First, incubation of either reagent with IleRS resulted in a rapid loss of enzyme activity. Both the tRNAIle aminoacylation and isoleucine-dependent isotopic ATP-PPi exchange activities were abolished in parallel by the labeling. Inactivation of IleRS by either reagent most probably proceeded through the formation of a Schiff base between the aldehyde group of ADP-PL or of tRNAIleox and amino groups of the protein. Second, the stoichiometries of labeling of IleRS by ADP-PL or tRNAIleox corresponded to 1 mol of reagent incorporated per mol of enzyme, consistent with the 1:1 IleRS:ATP or IleRS:tRNAIle active stoichiometries. Third, in the case of ADP-PL-labeling of IleRS, enzyme inactivation showed saturation behavior with respect to the reagent concentration, indicating that ADP-PL binds reversibly to the enzyme, prior to the covalent bond formation.
The plot of the fraction of inactivated IleRS versus increasing reagent concentrations indicated a half-effect concentration of ADP-PL in the inactivation process of about 0.17 mM, suggesting that the affinity of ADP-PL is comparable to that of ATP (Kd = 0.176 mM) [31]. Finally, intact tRNAIle or MgATP protected IleRS from inactivation by tRNAIleox, while incubation of the synthetase with MgATP prevented enzyme inactivation and labeling by ADP-PL. Altogether, these results suggest that labeling of IleRS by ADP-PL or by the oxidized 3'-adenosine of tRNAIle occurs specifically at the enzyme active centre.
The chymotryptic digestion of the [14C]tRNAIleox-labeled IleRS yielded a unique 603SKSIGN608 labeled peptide in the primary structure of the synthetase, with the 3'-oxidized adenosine covalently attached to Lys-604. In the case of ADP-PL-labeling of IleRS, the labeled residues were Lys-601 (fragment [601-604]) and Lys-604 (fragment [602-618]) (Fig. 2). Thus, altogether, the oxidized 3'-end of tRNAIle and the pyridoxal moiety of the ATP analog ADP-PL react with lysines 601 and 604 of the consensus sequence 601KMSKS605. This motif was first discovered at the active centre of E. coli MetRS, as a consequence of affinity labeling studies [3, 4, 6]. In fact, lysine-335 of the 332KMSKS336 sequence in E. coli MetRS was identified as the residue majorily labeled by the oxidized 3'-adenosine of tRNAfMet. It was proposed that Lys-335 could serve as a ligand for the CCA arm of tRNA through electrostatic interactions between a phosphate group of the nucleic acid and the ε-amino group [23]. Later on, Lys-335 in MetRS was found affinity labeled by the ATP analog adenosine triphosphopyridoxal (ATP-PL), suggesting that this residue might contact and help orientate the γ -phosphate of bound ATP in such a way as to facilitate pyrophosphate liberation as well [7]. Finally, Lys-335 was shown by site-directed mutagenesis to be involved through interaction with the γ-phosphate of ATP in the stabilization of the transition state during the synthesis of methionyl-adenylate [32]. Similarly, Lys-604 of IleRS may serve either to guide the tRNA CCA arm or to help adenylate formation by interacting with the phosphate chain of the ATP substrate.
This is most probable since, as already discussed, the binding site for the CCA arm of tRNA might be close to the site for the formation of aminoacyl-adenylate, since the 3'-end of tRNA has to react with the anhydride bond of the enzyme-bound aminoacyl-adenylate.
It should be noted that, Lys-604 of E. coli IleRS and the corresponding Lys-335 of E. coli MetRS are strictly conserved in the IleRS (Fig. 10) and MetRS families [33], respectively, as well as in the so called class 1 aminoacyl-tRNA synthetases, as expected for residues involved in the stabilization of the transition state.

Alignment of amino acid sequences of IleRS from different origins, around the identified labeled residues of E. coli IleRS.
The labeled amino acid residues covalently attached to the reactive substrate analogs used in this study are numbered (in bold) according to the numbering of the corrected IleRS amino acid sequence from E. coli reported here. Alignment of amino acid sequences of IleRS from Staphylococcus aureus (Sa), Thermus thermophilus (Tt) and Homo sapiens (Hs) with that of the same synthetase from E. coli around the identified labeled residues was achieved with CLUSTAL. Asteriks indicate identical residues, double dots and single dots indicate respectively highly and weakly conserved residues.
Since the covalent binding of the histidine-specific reagent DEPC to His-591 and His-905 of E. coli IRS, was accompanied by enzyme activity loss, it is likely that modification of His-591 situated at only a ten-residue distance from lysines-601 and -604 (Fig. 10) might be responsible for a steric hindrance which prevents the access of substrates to the catalytic site.
The Binding Site for L-isoleucine or for Non Cognate Amino Acids on E. coli IleRS
In the double-sieve editing mechanism proposed by Fersht [13, 14, 34], amino acids that are smaller than the correct one might be activated to the adenylated form, in the active site of a given synthetase, while amino acids larger than the cognate might be rejected. In support of this hypothesis, IleRS was previously shown to activate both isoleucine and valine to their adenylated form, while this synthetase could not efficiently misactivate methionine or leucine [35].
On the other hand, the double-sieve mechanism supposes that the products of misactivation of amino acids that are smaller than the correct one might be accepted in the editing site of the synthetase, while the cognate aminoacyl adenylate would be rejected from this site, in order to prevent its hydrolysis.
Interestingly, in the report by Nureki et al. [15], the crystal structure of IleRS from Thermus thermophilus solved at a 2.5 Å resolution shows two catalytic sites, one for aminoacylation with the substrate isoleucine, and one for editing of misactivated valine.
Actually, the substrate analog IBMK was found covalently attached to Cys-462 and Cys-718, in the primary structure of E. coli IleRS. Cys-462 was previously identified as the target of IBMK at the active center of E. coli IleRS [36]. The present study therefore confirms these reports, and at the same time designate Cys-462 as the binding site for L-isoleucine and/or the synthetic site where isoleucyl adenylate formation takes place.
With the exception of His-332 (or His-337), which was found labeled only by VBMK, the reactive analogs of L-valine and L-norleucine, two amino acids chemically similar to L-isoleucine were covalently attached to a set of four residues (His-394, Cys-462, His-478 and Cys-718), two of which (Cys-462 and His-478) belong to fragment [452-480] in the amino acid binding domain of the synthetase. Labeling of Cys-462 by VBMK and NleBMK might reflect that they are comparable in size or smaller than the substrate isoleucine, and that, consequently, they are well fitted to the isoleucine binding site, in agreement with the double-sieve editing mechanism [13, 14, 34]. The fact that the bromomethyl ketone derivative of phenylalanine which is larger than isoleucine could not react with Cys-462 suggests that this reagent which is larger than IBMK, is not in proximity of binding with Cys-462 of the isoleucine-binding site on E. coli IleRS, in agreement with the double-sieve hypothesis of Fersht [13, 14, 34].
Therefore, inactivation of IleRS by FBMK might be accounted for, by the covalent attachment of the reagent to His-478 which prevents the access of substrates to Cys-462 of the active site.
All the used bromomethyl ketone derivatives of non cognate amino acids (VBMK, NleBMK and FBMK) labeled in common His-394 (fragment [325-394]) in the editing domain of E. coli IleRS. In addition, VBMK specifically labeled His-332 (or His-337) in this domain (Fig. 10). Labeling of His-332 (or His-337) by VBMK but not by IBMK, NleBMK or FBMK might be explained as follows: (i) valine (Van der Waals volume : 105 Å3) is smaller than the substrate isoleucine (Van der Waals volume : 124 Å3), so that its product of misactivation, or the reactive analog thereof (namely VBMK) would be accepted in the editing site of the synthetase, as proposed by the double-sieve mechanism; (ii) in contrast, the adenylate form of the cognate isoleucine or of larger non cognate amino acids as well as their analogs (IBMK, NleBMK or FBMK) would be rejected from this site.
In conclusion, the fact that VBMK, NleBMK and FBMK label each Cys-462 and/or His-478, in the isoleucine-binding domain on one hand, and His-332 (or His-337) and/or His-394 in the editing site on the other hand, might be the reflect of the existence of two binding sites for non cognate amino acids on IleRS, in agreement with the reports by Nureki et al. [15] and Silvian et al. [16] for IleRS from T. thermophilus and S. aureus.
The Editing Site of E. coli IleRS
Comparison of fragment [322-397] with IleRS amino acid sequences (Fig. 10) revealed that His-332 is strictly conserved, while the neighbouring His-337 is only weakly conserved in these synthetases. By contrast, His-394 is not conserved at all in the IleRS family. These results suggest that histidines-332 and -337 might be of functional significance in the IleRS family.
Mutational analysis of the E. coli IleRS editing active site has demonstrated a crucial role for His-332 in the hydrolytic editing activity of the enzyme [37]. An H332A mutant IleRS from E. coli, referred to as H333A in Hendrickson et al. [37], was shown to exhibit the following properties: (i) H333A IleRS edites isoleucine, because it stimulates tRNAIle-dependent ATP hydrolysis in the presence of isoleucine; (ii) H333A IleRS deacylates Val-tRNAIle and Ile-tRNAIle at similar rates comparable to that of the wild-type enzyme with Val-tRNAIle as substrate; (iii) both activated isoleucine and valine, in the form of their respective adenylates, are translocated at similar rates by H333A from the active site to the editing center of the enzyme. These results suggest that H333A IleRS is altered in amino acid specificity for post-transfer editing. Affinity labeling of His-332 of IleRS by the bromomethyl ketone derivative of non cognate valine directed toward the editing active site is in agreement with previous work carried out with the site directed mutagenesis approach [37].
In a previous work, we have used affinity labeling [12] and protein modification [38] studies to identify amino acid residues at the synthetic and the hydrolytic editing active sites on E. coli ValRS. Among the amino acid residues of the editing active site of this synthetase, Lys-277, a residue strictly conserved from bacteria to human, corresponds to His-332 of E. coli IleRS [38] which plays a crucial role in editing ([37] and this work). Lys-277 of E. coli ValRS was also shown by site-directed mutagenesis, to be involved in the editing activity. Indeed, a point Lys277Ala (K277A) mutation yielded a mutant E. coli ValRS that presented the following properties: (i) the K277A mutant exhibited a posttransfer Thr-tRNAVal rate significantly lower than that observed for the wild-type enzyme; (ii) the K277A substitution altered amino acid discrimination in the editing site, resulting in hydrolysis of the correctly charged cognate Val-tRNAVal ; (iii) significant amounts of mischarged Thr-tRNAVal were produced by the K277A mutant, and not by the wild-type ValRS.
Primary structure alignment of the conserved editing region within the CP1 domain of IleRS, ValRS and LeuRS, reveals that His-332 of E. coli IleRS and the corresponding Lys-277 of E. coli ValRS, match with His-319 of T. thermophilus IleRS and with Lys-270 of T. thermophilus ValRS [38]. Moreover, in the tRNA-complexed T. thermophilus IleRS [15] and ValRS [39] 3-D structures, the editing domain CP1 containing His-319 (IleRS) and Lys-270 (ValRS), is arranged in a similar position. Thus, His-332 and His-319 of IleRS from E. coli and T. thermophilus, respectively, as well as Lys-277 and Lys-270 of ValRS from E. coli and T. thermophilus, respectively, which play a crucial role in editing in the CP1 domain, are located at similar positions in the amino acid sequences [38] and in the 3-D structures of these synthetases [15, 39].
The involvement of nucleophiles in the CP1 editing domain of the aminoacyl-tRNA synthetases which are concerned with editing has been extensively discussed [37-42]. However, their precise role in the editing active site remains to be elucidated. For example, in the crystal structure of IleRS from Thermus thermophilus solved at a 2.5 angstroms resolution, six residues (Thr-228, Thr-229, Thr-230, Thr-233, Asn-237 and His-319) are located in close proximity, in a way that is reminiscent of the catalytic triads of hydrolases [15]. However, the nucleophile His-319 of Thermus thermophilus IleRS is not conserved in the ValRS and LeuRS amino acid sequences, suggesting that the involvement of a mechanism analogous to the charge relay system in the editing site of the ValRS, IleRS and LeuRS families is less probable. On the bases of the data of the present work on Escherichia coli IleRS, and of the qualitative comparative labeling of Escherichia coli ValRS with reactive amino acids analogs [12, 38], we propose that the role of nucleophilic residues in the editing active site of the synthetases consists of merely helping to deprotonate the catalytic editing residues, in order to make them capable of attacking the carbonyl group of the ester of misactivated or misacylated non cognate amino acids. This hypothesis is supported by the observation by Edsall and Wyman [43], that situation of a nucleophile at a two-residue distance from another one may increase the nucleophilic character of each of them, by lowering the pKa values by up to 2 pH units. The nucleophilic attack resulting in the hydrolysis of the ester bond might insure an accurate translation process, by preventing misincorporation of non cognate amino acids into proteins. Following this nucleophilic attack, the covalent bond formed between the catalytic editing residues and the rejected non cognate amino acids might be difficult to detect with current analytical biochemistry tools because of the low mass of those amino acid adducts with regard to the whole enzymes masses. Work is in progress, with new generation mass spectrometry equipments, to detect such post-editing covalent protein modifications.
CONFLICT OF INTEREST
This work was supported by grants from CNRS and the Laboratoires GLAXO (Les Ulis, Courtaboeuf, France).
ABBREVIATIONS
REFERENCES
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]

