All published articles of this journal are available on ScienceDirect.

COVID-19: Mechanisms of the Antiviral Activities of Selective Antibiotics Targeting the Human 80S Ribosome
Authors Info & Affiliations
Abstract
Background:
The majority of scientists, physicians, and healthcare professionals were trained with the paradigm: “antibiotics are for bacteria only !”, because they misunderstood the definition of the ribosome targeting antibiotics. In the context of the current worldwide COVID-19 pandemic, it might be useful to recall as precisely as possible the definition of the word antibiotic and provide evidence that some classes of antibiotics could offer excellent means to counteract viral infections via specific mechanisms.
Methods:
Molecular modeling and docking studies were used, as well as the tRNAox labeling reaction of the ribosomal protein eL42 in situ on human 80S ribosomes to demonstrate that cycloheximide and its thiosemicarbazone analogues bind to the catalytic Lys-53 residue of the human large subunit ribosomal protein eL42.
Results:
Comparison of the binding sites for Cycloheximide (CHX) and Sparsomycin (SPS) on the evolutionarily conserved E. coli bL12 and S. cerevisiae eL42 by means of molecular modeling and docking studies showed that: (i) SPS binds in proximity to the catalytic Lys-65 residue of the GANK motif of rp bL12 and to the catalytic Lys-55 residue of the GGQTKP motif of rp eL42; (ii) CHX failed to bind to the GANK motif, while the glutarimide moiety of SPS and CHX was found to make contact with Lys-55 of the GGQTKP motif of rp eL42.
Conclusion:
In this report, we demonstrate that cycloheximide and its thiosemicarbazone analogues are capable of inhibiting the human 80S ribosomes selectively through their binding to the ε-amino group of the side chain of Lys-53. As a consequence, these small-molecule inhibitors of translation are susceptible to exhibit antiviral activities by preventing the human ribosomes of the SARS-CoV-2 infected cells from synthesizing the viral proteins and enzymes.
1. INTRODUCTION
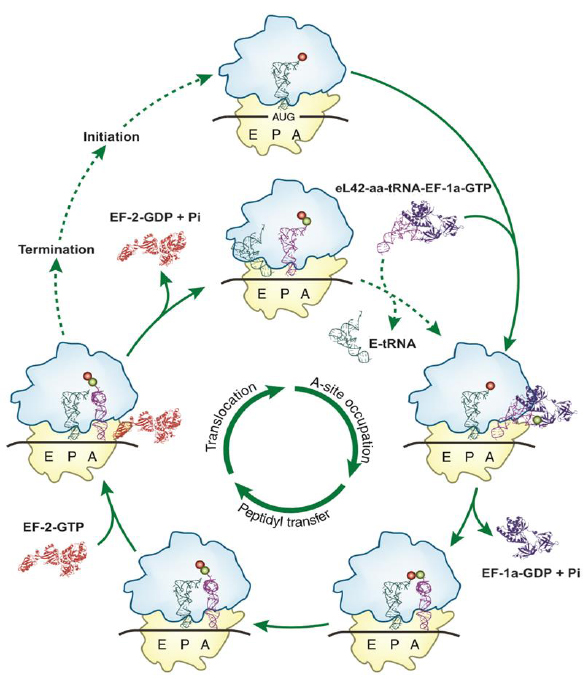
Considering the urgent need for effective treatment to combat the betacoronavirus SARS-CoV-2 responsible for the COVID-19 pandemic, the following observations and demonstrations have been made. First, it is most probable that the discovery (if any) of clinically approved vaccines against SARS-CoV-2 would not make it optional to search for specific and effective therapeutic drugs for COVID-19, as many people are susceptible to be concerned by contraindications. Therefore, in addition to designing and developing vaccines for SARS-CoV-2, repositioning of old drugs such as antibiotics and the development of new drugs are also needed. Second, it is widely accepted that viral infections cannot be cured by antibiotics whereas bacterial infections can as the virus has no protein synthesis machinery (the normal target for antibiotics) to produce from its RNA genome, the viral proteins and enzymes essential to its replication and/or to the assembly of viral particles. As a consequence, in the SARS-CoV-2 infected host cells, the main target for the therapeutically useful drugs is the human protein synthesis machinery built up with the ribosomes, because the virus is forced to rely on the infected host cell to synthesize new proteins that it does not need, but are needed for the virus to spread. In this regard, it is generally accepted that antibiotics that block the translation process in mammalian cells have potential as therapeutics for diseases such as cancer, inflammation or COVID-19. However, there is a long-standing debate on whether or not antibiotics can be used to cure viral infections such as COVID-19. To address this question, is it enough for physicians to know that a particular antibiotic is effective against a given disease? Obviously not, because for the intelligent and rational use of antibiotics in the treatment of any disease, the physician must have detailed knowledge of how antibiotics work. With this type of knowledge, the right choice or combination can be made to guarantee optimum success in the treatment. In the present report, we have revisited the definition of the antibiotics and identified the large subunit ribosomal protein eL42 as a novel target for eukaryote specific antibiotics and their analogues in the SARS-CoV-2-infected host cells, with the goal of developing specific and effective therapeutic drugs for COVID-19. In fact, recent studies from our group have demonstrated that the large subunit ribosomal proteins bL12 and eL42 assist catalysis by correctly positioning the incoming aminoacyl-tRNA in the A-site of 70S or 80S ribosomes of bacteria and eukaryotic cells, respectively [1-6] (Fig. 1). Finally, it had been previously reported that rp eL42 of the human 80S ribosome is the target for small-molecule inhibitors of translation. Accordingly, molecular modeling and docking studies have been used to compare the binding sites for small-molecule inhibitors on eL42 and bL12.
2. MATERIALS AND METHODS
2.1. Molecular Modeling and Docking Studies
Small molecule structures were obtained from PubChem and converted to the appropriate formats for further elaboration in different softwares using the Babel program. OpenBabel, JoeLib, and ChemMimeR were employed to calculate chemical descriptors necessary to evaluate druggability (e.g., “Lipinski’s rule of 5”). Structure alignment of identified candidate molecules (sparsomycin, cycloheximide) was performed to detect potential similarities using the Escan software arising from the on-line 3DMSS. Homology modeling was performed classically by mutating key side-chain residues into the appropriate ones, and the resulting structure was energy-minimized down to the expected gradient. Three softwares were employed to verify the quality of the resulting final structures, namely Verify-3D, QMEAN (Qualitative Model Energy Analysis), and PROCHECK. For docking small molecules to macromolecules, we used Autodock with great success.
2.2. Crosslinking with tRNAox of Human 80S Ribosomes
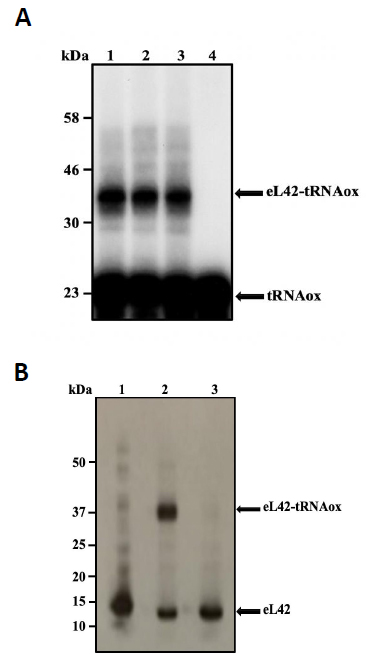
Human 40S and 60S ribosomal subunits with intact rRNAs were isolated from unfrozen human placenta. Prior to use, the subunits were reactivated by incubation in buffer Tris·HCl 50 mM (pH 7.5) containing KCl (100 mM), MgCl2 (10 mM) and EDTA (0.5 mM) at 37°C for 10 min. The 80S ribosomes were obtained by association of the reactivated 40S and 60S subunits taken in equimolar ratio. The ribosomes (0.5 μM) were incubated at 37 °C in buffer A (50 mM Tris–HCl, pH 7.5, 100 mM KCl, 10 mM MgCl2 and 0.5 mM EDTA) containing 5 μM of the dodecaribonucleotide GAA UUU GAC AAA as an mRNA, in a total volume of 25 μl. The labeling reaction was initiated by the addition of 10 μM [32P] tRNAAspox and 5 mM sodium cyanoborohydride (NaBH3CN), and the incubation continued for 1 h. After the termination of the tRNAox-labeling reaction, the incubation mixtures were quenched by the addition of 1 μl of 0.5 M sodium borohydride (NaBH4) freshly prepared in 50 mM phosphate buffer (pH 7.5). 20 μl portions of the NaBH4-quenched samples were applied onto a 10% polyacrylamide gel which was run by SDS-gel electrophoresis. The gels were dried under vacuum and subjected to autoradiography.
3. RESULTS AND DISCUSSION
3.1. Defining Ribosome Targeting Antibiotic Type Molecules in the Context of Human Diseases
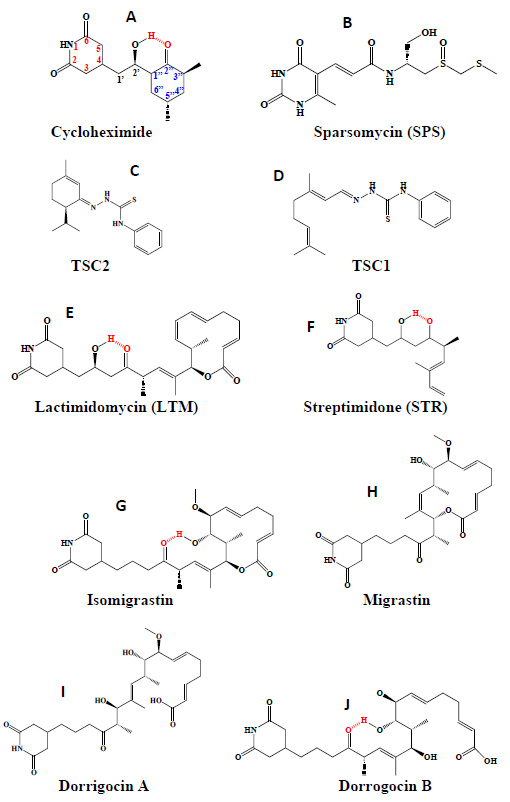
Given that, in the context of the worldwide COVID-19 pandemic, the word antibiotic is currently frequently misused, it might be useful to recall as precisely as possible its definition. Antibiotics are chemical weapons that species create over evolutionary times to seclude a niche in which they can develop and grow safely in their zone of exclusion of other competing species. Men, either from natural resources or human engineering, created a realm of such chemical weapons mainly against bacteria, parasites and insects. So far, however, our warfare against virus species has been hampered by the lack of such chemical weapons, and the coronavirus disease 2019 (COVID-19) outbreaks found us unarmed. The purpose of this paper is precisely to broaden the scope of antibiotics. Most scientists were trained with the paradigm: “antibiotics are for bacteria only!”. We now provide evidence that some classes of antibiotics could offer excellent means to counteract viral infections via the mechanisms exposed in this paper. In principle, an antibiotic is an antibacterial agent used to treat or prevent bacterial infections. In the usual medical sense, an antibiotic is produced naturally by a microorganism with the goal of fighting another microorganism. These two canonical criteria are necessary to the definition of a given antibiotic. Many antibiotics that are widely used in clinical medicine act at different functional sites on the ribosome, where they inhibit protein synthesis [7]. Most of the antibiotics selectively or preferentially inactivate bacterial or archaeal ribosomes, in accordance with the aforementioned definition. However, those referred to as universal antibiotics are capable of universally inhibiting the ribosomes in all kingdoms of life. Therefore, they are equally capable of inhibiting the translation process in mammalian cells and can be considered as potential therapeutics for diseases such as cancer, inflammation or COVID-19. Examples of universal antibiotics include macrolides and tetracyclines. In sharp contrast to the majority of antibiotics, Cycloheximide (CHX) (Fig. 2) is specific only for the ribosomes of eukaryotic cells or fungi. It is a naturally occurring compound produced by the bacterium Streptomyces griseus. CHX exerts its effects by inhibiting eukaryotic translational elongation, thus blocking protein synthesis. In general, the glutarimide antibiotics, of which CHX is the best known member, are derived from various species of Streptomyces, and are known to inhibit protein synthesis and to exert antitumor and fungicidal actions. Given that an antibiotic is produced by a microorganism (first canonical criteria cited above), with the goal of fighting another microorganism (second canonical criteria), the fact that CHX is capable of inhibiting protein synthesis in eukaryotic cells, not in bacteria, raises the question of whether this compound can be named antibiotic. However, since CHX is produced by the bacterium Streptomyces griseus (first canonical criteria), it is generally considered an antibiotic (Table 1). Finally, due to significant toxic side effects, CHX is generally used only in in vitro biomedical research and is not suitable for human use as a therapeutic compound. As discussed in another section, we have used the structure-assisted drug design approach to synthesize small-molecule inhibitors analogous to CHX [8]. Another example of glutarimide antibiotic is sparsomycin (SPS) (Fig. 2), a compound initially discovered as a metabolite of the bacterium Streptomyces sparsogenes, which binds to the Peptidyl Transferase Center (PTC), the active site of the bacterial ribosome located on the 50S subunit, and inhibits protein synthesis [9, 10]. SPS was also formerly found to be an antitumor agent. It is capable of inhibiting protein synthesis in both eukaryotic cells and bacteria [9, 10], which raises the question of whether or not this compound can be named antibiotic. However, as discussed above about CHX, this compound is produced by a bacterium (Streptomyces sparsogenes), and might be therefore considered as an antibiotic Table 1. It remains that neither CHX, nor SPS is usable as a therapeutic compound in humans. In the present report, we have used molecular modeling and docking studies to compare the binding sites on the ribosomes of CHX, or SPS or small-molecule inhibitors analogous to CHX, for the following reasons: (i) SPS inhibits protein synthesis in both eukaryotic cells and bacteria, while CHX is capable of inhibiting protein synthesis in eukaryotic cells, not in bacteria. Since these structurally similar glutarimide antibiotics (Fig. 3) bind to the PTC of the ribosomes, it is most probable that they target evolutionarily conserved ribosomal functional sites; (ii) we have recently demonstrated that the large subunit ribosomal proteins bL12 and eL42 assist catalysis at the PTC by correctly positioning the incoming aminoacyl-tRNA in the A-site of 70S or 80S ribosomes of bacteria and eukaryotic cells, respectively [1-5]. In addition, ribosomal proteins bL12 and eL42 were recently shown to be evolutionarily conserved at the tRNA-CCA binding site of the ribosomes [3]; (iii) it was previously reported that rp eL42 of the archaeal or eukaryal ribosomes is the target for small-molecule inhibitors of translation such as CHX [11, 12].
| Antibiotics | Class | |
| B/E | Tetracycline | Tetracycline |
| B | Streptomycin | Aminoglycoside |
| B | Paramomycin | Aminoglycoside |
| E | Geneticin G418 | Aminoglycoside |
| Inhibitors of the Large Ribosomal Subunit | ||
| B/E | Sparosomycin | Glutarimide |
| E | Cycloheximide | Glutarimide |
| B/E | Erythromycin | Macrolide |
| B/E | Azithromycin | Macrolide |
3.2. Protein Synthesis on the Ribosome: New Insights into the Mechanism of Translation Elongation in Bacteria and Eukaryotic Cells
As discussed in a previous report, whether they belong to the SARS-CoV-2 virus or to the infected human host cell, all the enzymatic or non enzymatic proteins are synthesized by the human protein synthesis machinery centered on the ribosome [13]. Thus, on one hand, the human 80S ribosome will use the viral RNA as a template to translate all the enzymes and proteins necessary for the multiplication of the virus and for the assembly of viral particles. On the other hand, the human 80S ribosome will use human mRNA as a template to translate the cytokines that witness the viral infection. The translation process has been a subject of controversy for a long time. In the post-crystal-structure era of the studies on the ribosome, starting from the year 2000, the view of ribosomal peptidyl transfer during translation elongation was that the ribosome was a ribozyme (i.e. a catalytic RNA) and that ribosomal proteins were not involved in the catalysis of peptide bond formation [14-16]. This view originated from the fact that the three-dimensional structure of the 50S ribosomal subunit of the archaeon Haloarcula marismortui (Hma) revealed a void of protein electron density in a radius of 18 angströms of the PTC [14, 15]. However, in sharp contrast to the ribozyme-catalyzed peptidyl transfer mechanism of the ribosome, our group has recently provided new insights into the mechanism of peptide bond formation by demonstrating that the large subunit ribosomal proteins bL12 and eL42 assist catalysis by correctly positioning the incoming aminoacyl-tRNA in the A-site of 70S or 80S ribosomes of bacteria and eukaryotic cells, respectively [1-5]. Fig. (4A-C) show the model of the 3-D structure of human rp eL42 alone or in a complex with tRNA, as well as the functional domains of this protein including the tRNA-CCA binding site, the zinc finger domain and the Nucleotide-Binding Domain 2 (NBD2) [17]. Ribosomal proteins bL12 and eL42 were recently shown to be evolutionarily conserved at the tRNA-CCA binding site of the ribosomes [3]. First, these two proteins have comparable apparent molecular weights of about 12,000 Da. Second, they were shown to contact the CCA-arm of P-site bound tRNA [3]. Third, they display primary structure similarities, with an average of 44% of identities and conservative replacements [3]. In particular, the 62GANK65 motif of bL12 matches with the 49GGQTK53 motif of eL42 [3] (Fig. 5A). Fourth, it has been recently demonstrated that Lys-65 of E. coli bL12 and Lys-53 of human (Lys-55 in Yeast) eL42 exhibit an abnormally low pK (6.9), as compared with the one that is normally found for the side chain of a lysine residue (pK 10.5) [2-5]. Fifth, the GANK and GGQTK motifs occupy respectively comparable positions in the 3-D structures of E. coli bL12 and yeast eL42 [3] (Figs. 5B, C). Since the eukaryote specific rp eL42 protein is strongly conserved in eukaryotes and archaea, these data argue for evolutionary conservation of these two proteins located at the tRNA-CCA binding site on eubacterial, eukaryal and archaeabacterial ribosomes. The mechanism for peptidyl transfer reaction supporting peptide bond formation is shown in Fig. (6). Taking into account the pK value of 6.9 for Lys-65 of E. coli bL12 and Lys-53 of human (Lys-55 in Yeast) eL42, we had proposed that, at neutral pH, on one hand, the fraction (50%) of protonated, positively charged εNH+3 side chain of each residue establishes ionic bonds with phosphate groups of the polyanionic tRNA molecule. On the other hand, the remaining 50% fraction of unprotonated εNH2 withdraws a proton from the α-NH3+ group of the aa-tRNA positioned at the A-site. The latter reaction generates a nucleophilic α-NH2 group which subsequently performs the catalysis of peptide bond formation by a nucleophilic attack on the electrophilic carbonyl group of the peptidyl-tRNA positioned at the P-site (Fig. 6) [2, 4]. In conclusion, Lys-65 of E. coli bL12 and Lys-53 of human (Lys-55 in yeast) eL42 can be considered as targets for small-molecule inhibitors capable of inhibiting the ribosome of a host human cell infected by SARS-CoV-2 virus, as well as that of an opportunistic pathogenic bacterium present in the respiratory tract [2, 4].
3.3. Selective Inhibition of the Human 80S Ribosomes: Structural basis for the Inhibition of the Human 80S Ribosomes by Cycloheximide and the Glutarimide Antibiotics
As discussed above, in a host cell infected by the SARS-CoV-2 virus, the main target for the therapeutically useful drugs is the human ribosome [13]. Therefore, it is preferable to selectively inhibit the human ribosome in order to prevent it from synthesizing the proteins and enzymes essential to the replication and the assembly of the virus. Selective inhibition of the human 80S ribosomes is a task for cycloheximide (CHX) (Figs. 2 and 3). As discussed above, the newly discovered rp eL42 from human 80S ribosomes contributes directly and actively through its catalytic Lys-53 residue (Lys-55 in yeast and Lys-51 in Hma) to the catalysis of peptide bond formation at the elongation step of translation [2] (Figs. 1 and 6). Interestingly, in the crystallographic structure of S. cerevisiae 80S ribosomes or of the 50S subunit of Haloarcula marismortui, the antibiotic CHX and its analogues (Fig. 2) of the glutarimide class (which are known to abolish the activity of the ribosomes), as well as other anticancer drugs, were shown to target the eL42 protein by making contact with the catalytic residue Lys-55 of yeast rp eL42 (Lys-53 in human and Lys-51 in Hma) (Fig. 7A) [11, 12, 18, 19]. These data are consistent with the importance of rp eL42 through its ribosomal role in translation or its extra ribosomal role in the development and progression of cancer.



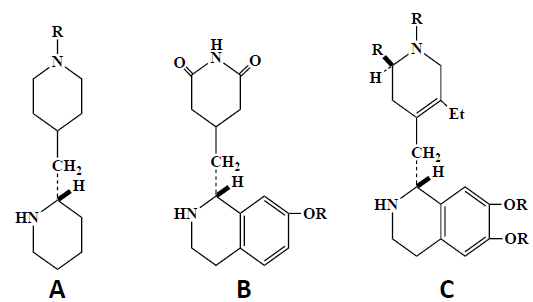
All the glutarimide type compounds feature a common pharmacophore, i.e., a glutarimide heterocycle and a 3-hydroxyethylketone moiety linked together by a variable short methylene sequence (Fig. 2). A pharmacophore is a trait that characterizes a class of drugs, and it contains coded in a group of linked atoms a message that is conveyed by small molecules to a therapeutic biological target macromolecule. In this common structural template, there is this 3-hydroxyethylketone template bearing an internally hydrogen-bonded motif (Fig. 2), this pseudo-hexacycle plays an important role in orienting correctly the two external cyclic structures and therefore creates an assembly that is recognized by the biological target. Additionally, these two peripheral cyclic structures create a hydrophobic shield which protects this internal hydrogen bond from disruption by water intrusion in biological media. This hydrophobic shield is also of major importance for drug transport, as somehow it “buries” two polar moieties (carbonyl ketone and hydroxyl groups) in a lipophilic continuum and therefore facilitates hydrophobic membrane crossing. This phenomenon has been called hydrophobic collapse [20]. Interestingly, the intramolecularly hydrogen-bonded conformation of CHX and analogues (Fig. 2) appears to be indispensable for inhibition of translation and for the antitumor activity [12]. Thus, all active analogues (Fig. 2) containing the hydrogen bond and a 12-membered macrocycle (LTM and isomigrastatin) were previously shown to inhibit cell proliferation and the translation process [12]. By contrast, none of the migrastatin-based 14-membered macrocycles and none of the dorrigocin-based linear isomers (Fig. 2) showed any activity [12]. This result would reflect steric effects since the 14-membered macrocycle is susceptible to create significant hindrance at the eL42 binding pocket on the eukaryal ribosome, which would be correlated with a decrease in inhibition activity. Despite the presence of a hydrogen bond in its structure, dorrigocin B analogue has no effect [12], which is in agreement with the importance of the steric effects. Another interesting analogue of CHX is the ipecac alkaloid emetine (Fig. 8) [21]. Despite the difference in their chemical structure, emetine presents conformational similarities to CHX and the glutarimide antibiotics and have similar effects on protein synthesis [21]. For example, emetine inhibits protein synthesis in mammalian cells, in the yeasts, as well as in plants, but not in the bacteria [21], similarly to CHX, suggesting that these drugs might bind to the same or proximal site(s) on the 80S ribosomes of eukaryotic cells. Representative compounds A-C contain the minimal structural requirements for the inhibition of protein synthesis based on the analogy between the emetine (C type) and the glutarimide antibiotics (B type) [21]. The hydrogen-bonded quasi ring of CHX corresponds to the ring of emetine [21].
3.4. Structure-Assisted Drug Design Approach for the Synthesis of Small-Molecule Inhibitors Analogous to Cycloheximide: Docking Studies
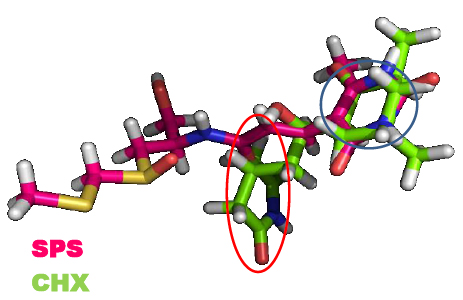
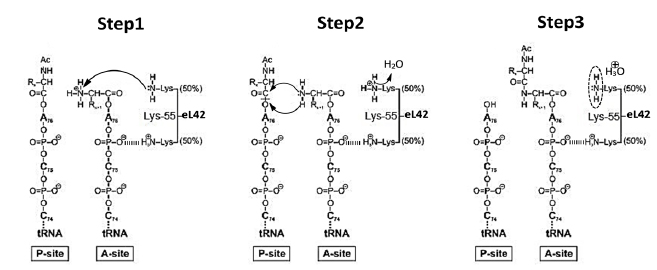
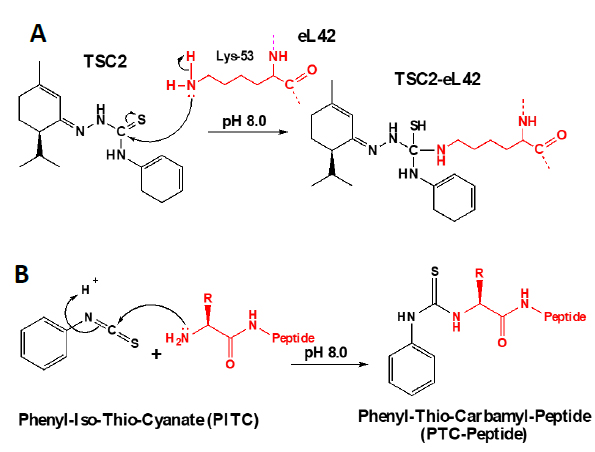
In addition to the catalytic role of the Lys-55 residue of eL42 [2], another argument which is in favor of the involvement of the 51GGQTKP56 motif in the activity of the yeast 80S ribosome is that several yeast strains are known to exhibit resistance mutations, one of which consisting in the replacement of the Pro-56 residue by Gln-56 [22, 23]. This mutation renders the yeasts resistant to CHX [22, 23]. The fact that CHX inhibits protein biosynthesis by binding to Lys-55 of eL42, while the mutation P56Q renders some yeast strains resistant to cycloheximide (CHX), strongly supports the involvement of these two adjacent amino acid residues (Lys-55 and Pro-56) in the binding and in the inhibitory effect of CHX on the activity of the ribosome. As discussed above, CHX was found to be a poisonous drug for mammals. Therefore, we have used the structure-assisted drug design approach to synthesize small-molecule inhibitors analogous to this glutarimide antibiotic. These molecules are the thiosemicarbazones (TSCs) [8] (Fig. 2) that were recently shown to have a potent antiproliferative effect on tumor cell lines similarly to CHX ([8] and unpublished data). We have used molecular modeling and docking studies to compare the binding sites for CHX and thiosemicarbazone2 (TSC2) in Fig. (8) on eL42. As shown in Fig. (7B), the glutarimide moiety of CHX is hydrogen-bonded to the side chain of the Lys-55 residue of S. cerevisiae eL42, as deduced from the crystal structure of the CHX:ribosome complex [11]. Similarly, the molecular modeling and docking studies (Fig. 9) show that the thiocarbonyl group of TSC2 lies in the proximity of binding with the side chain of the Lys-55 residue of S. cerevisiae eL42. However, in contrast to CHX, the thiocarbonyl group of TSC2 was shown to covalently react with the amino group of the side chain of Lys-53 of rp eL42. The hydrogen bond formed between the glutarimide moiety of CHX and the side chain of the Lys-53 residue of human eL42, was confirmed by the fact that CHX could not compete with the reactive tRNA dialdehyde derivative (tRNAox) for the covalent reaction with the ε-amino group of the side of Lys-53. In other words, the presence of CHX in the incubation mixture of the crosslinking reaction did not prevent the covalent reaction between tRNAox and the Lys-53 residue (Fig. 10A) By contrast, the fact that tRNAox failed to form a Schiff base and to crosslink with the Lys-53 residue of human rp eL42 suggests that TSC2 covalently reacts with this residue inside the human 80S ribosome [8] and (Fig. 10B). The latter reaction (Fig. 11A) is reminiscent of the well-known Edman degradation reaction where the free NH2 group of the amino-terminal residue of a protein (or peptide) covalently reacts with the Edman reagent phenylisothiocyanate (PITC) (Fig. 11B). Similarly, TSC2 covalently reacts with Lys-65 of rp bL12 residue inside the E. coli 70S ribosome (unpublished data), suggesting that this CHX analogue behaves similarly to a universal type antibiotic. Altogether, the data collected in the present report suggest that the TSC molecules (Fig. 2) would be capable of inhibiting selectively the 80S ribosomes of the human host cells infected by SARS-CoV-2 through a covalent reaction between their thiocarbonyl group and the ε-amino group of the side of Lys-53. This covalent reaction would prevent the human 80S ribosomes from synthesizing the viral proteins and enzymes. The TSC molecules are now ready to undergo clinical trials.
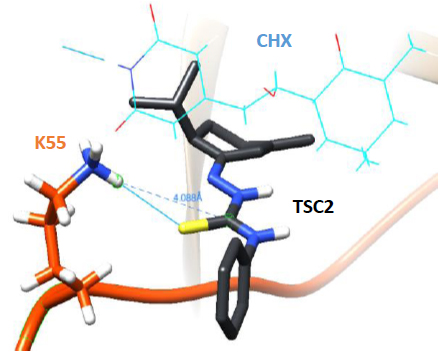
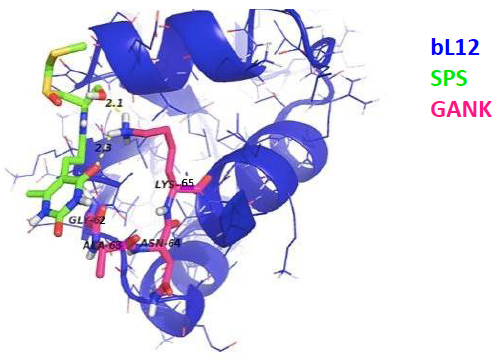
3.5. Comparison of the Binding Sites for Cycloheximide and Sparsomycin on E. coli bL12 and S. cerevisiae eL42
As discussed above, the newly discovered rp eL42 (formerly L42A or L42AB in yeast, L36a or L36a-like in human, or L44e in archaea) from human 80S ribosomes is indispensable for the activity of the eukaryal ribosomes and is likely to represent a target for small-molecule inhibitors in the human host cells infected by SARS-CoV-2. In fact, we have recently demonstrated that rp eL42 controls the rate of protein biosynthesis in health and in disease through its ribosomal and extra ribosomal functions. In health, the ribosomal function of rp eL42 consists of sustaining normal cell growth by catalyzing the elongation step of translation [1, 2, 5, 17]. In disease, we have recently demonstrated that one extra ribosomal function of rp eL42 might be related to abnormal cell growth characteristic of the proliferation of cancer cells [24]. In the coronavirus disease, as discussed above, rp eL42 (using the viral genome bound to the human 80S ribosomes in the infected host cells) is responsible for the synthesis of all viral proteins and enzymes, including the RNA-dependent RNA polymerase (RdRp), the RNA replicase which is primarily responsible for the multiplication of the SARS-CoV-2 virus [25]. Finally, the following observations and previous results need to be taken into consideration: (i) cycloheximide (CHX) inhibits protein synthesis in eukaryotic cells, not in bacteria, while sparsomycin (SPS) is capable of inhibiting protein synthesis in both eukaryotic cells and bacteria; (ii) both CHX and SPS are known to inhibit translation elongation at the PTC of the ribosomes; (iii) interestingly, as discussed above, in the crystallographic structure of S. cerevisiae 80S ribosomes, CHX and its analogues (Fig. 2) of the glutarimide class were shown to target the eL42 protein by making contact with the catalytic residue Lys-55 of rp eL42 (Fig. 7A) [11]. However, to our knowledge, the binding site of SPS had not so far been identified precisely on the ribosomes of eukaryotic cells. Therefore, it would be of great interest to compare the binding sites for CHX and SPS on S. cerevisiae 80S ribosomes by means of the docking studies; (iv) as discussed above, ribosomal proteins bL12 and eL42 were recently shown to be evolutionarily conserved, because they contact the CCA-arm of P-site bound tRNA [3] and display primary structure similarities, with an average of 44% of identities and conservative replacements [3]. In particular, the 62GANK65 motif of bL12 was shown to match with the 49GGQTK53 motif of eL42 [3] (Fig. 5A), while the catalytic residues Lys-65 of E. coli bL12 and Lys-53 of human (Lys-55 in yeast) eL42 were found to exhibit an abnormally low pK (6.9) and to assist catalysis on bacterial and eukaryal ribosomes, respectively [2-5]. Considering these data, we predicted that SPS would target the bL12 protein by making contact with the catalytic Lys-65 residue. To test this prediction, we have performed the docking of SPS on E. coli rp bL12. As a control, CHX would fail to bind to E. coli rp bL12, because this antibiotic is specific for eukaryotes only. As shown in Figs. (12 and 13), SPS binds in close proximity to the catalytic Lys-65 residue of the GANK motif of rp bL12. By contrast, CHX failed to bind to this motif, while the glutarimide moiety of SPS and CHX were found to make contact with Lys-55 of the GGQTKP motif of rp eL42, as expected.

3.6. Mechanisms of the Antiviral Activities of Selective Antibiotics Targeting the Human 80S Ribosome
As discussed in a previous report from our group, the macrolides and the tetracyclines represent universal antibiotics capable of universally inhibiting the ribosomes in all kingdoms of life [13]. Therefore, they are potentially effective therapeutics for diseases such as COVID-19. On one hand, the macrolides bind in the upper part of ribosomal tunnel, where they inhibit protein synthesis by interfering with the progression of the nascent peptide [13]. Considering that all proteins in the living cells are built up with the same common 20 or so amino acid residues, we assumed that the tunnel whereby their 1-dimensional structure exits from the ribosomes should be of comparable size and shape from top to bottom in all kingdoms of life. On the other hand, the tetracyclines are inhibitors of translation elongation which specifically prevent binding of tRNA to the A-site by interfering with the decoding site of the 30S ribosomal subunit [13]. Given that the anticodon of all tRNAs consists in trinucleotides complementary to 15 angström-long trinucleotide platforms (A-site codons) on the mRNA, the latter are expected to be universally conserved in all kingdoms of life. In conclusion, the protein exit tunnel and the A-site platform represent universally conserved functional sites and are likely to bind similarly the macrolides and the tetracyclines, respectively, in all kinds of ribosomes, the 70S as well as the 80S type, with comparable affinity. Accordingly, the antiviral activities of the macrolides and the tetracyclines consist in inhibiting the ribosomes of the SARS-CoV-2 infected human host cells in order to prevent them from synthesizing the viral proteins and enzymes [13]. Since rps bL12 and eL42 and their catalytic Lys-65 and Lys-53 residues are evolutionarily conserved at the tRNA-CCA binding site of the ribosomes in all kingdoms of life (eubacteria, eukaryotes and archaebacteria), we propose that the glutarimide antibiotics that target these catalytic residues represent a novel class of universal antibiotics. These glutarimide-type universal antibiotics and their thiosemicarbazone analogues bind to the catalytic Lys-53 residue of the human rp eL42. This binding is well known to cause the complete inactivation of the ribosome ([12] and unpublished data), followed by cytotoxicity leading to the death of cancer cells, for example [12, 26, 27]. The cytotoxic power of CHX is due to the fact that the catalytic residue Lys-53, its binding site on rp eL42, is the most important residue that supports Life through the synthesis of proteins and enzymes that are the support of Life, as well as of all the biomolecules themselves synthesized by these enzymes. Therefore, this very special molecule is intimately linked to the pathway of Life and Death of eukaryotic cells and archaebacteria. Here is the proof that the Lys-53 catalytic residue is essential for Life: our group has recently carried out on Schizosaccharomyces pombe (S. pombe) cells the site-directed mutagenesis of the gene encoding the ribosomal protein eL42, and we replaced the Lys-55 residue of yeast eL42 (corresponding to Lys-53 in human eL42) by a glutamine or a glutamic acid. The result is that the ribosome thus mutated is completely inactive and that this mutation is lethal for the yeast cells. Taking into account a molecular weight of 3.3 x 10+6 Da for the eukaryal ribosome and the presence of ribosome-bound translation factors, we have calculated that the human ribosome-centered protein synthesis machinery is comparable to a car engine made up of about 20,000 parts arranged in a specific order. Of these numerous parts, the fact that the presence of only one defective part (mutant Lys53Gln), out of 20,000, causes the death of the entire molecular engine is obviously of utmost spectacular. Note that this catalytic Lys-53 residue of rp eL42 also manufactures all the proteins and enzymes for SARS-CoV-2 responsible for the COVID-19 pandemic. Therefore, this lysyl residue of rp eL42 is a likely novel target for the glutarimide antibiotics, and for therapeutically useful derivatives thereof, to combat the betacoronavirus SARS-CoV-2 responsible of the COVID-19 pandemic. Interestingly, the antiviral spectrum of CHX against several DNA and RNA viruses have been evaluated. CHX showed strong inhibitory activities against several RNA viruses such as HIV-1, influenza viruses, coxsackie B virus, enterovirus (EV71) and several DNA viruses such as HSV and HCMV [28]. The Structure–Activity Relationship (SAR) of CHX and derivatives also has been established. The hydroxyl group at C-2’ and carbonyl group at C-2” of CHX (Fig. 2) are essential for its antiviral activity, and modification to these groups results in severe reduction or even total loss of antiviral activities. Since these groups were also previously shown to be essential to the cytotoxic effect of CHX [29], it is most probable that both antiviral and cytotoxic activities of CHX and its derivatives proceed by their binding to the catalytic Lys-53 residue of rp eL42, leading to the complete inactivation of the human ribosome. Another ribosomal protein located in close proximity to eL42 has also been implicated in CHX resistance. This rp is uL15 (formerly L28 in yeast or L27a in mammals). It is interesting to note that uL15 contains a 36GGQ38 motif in the peptide 30GGRGMAGGQHHHR42 involved in the binding of CHX, and a Q38E transition was found to be responsible for the resistance [30]. Interestingly, replacement in rp uL15 of the His-39 residue (adjacent to Gln-38) by Ala or Thr impairs the activity of human ribosomes [31]. Since rp uL15 is universally conserved in all kingdoms of life, these data strongly agree with early biochemical studies that had demonstrated that peptidyl transferase is an enzyme with an ionizable functional group of pKa 7.2, which was supposed to be a catalytic His residue [32, 33]. However, this residue has not been identified so far. Altogether, this result as well as previously reported data from our group demonstrate that not only the ribozyme model of the ribosome is disproved by the involvement of the eukaryote specific rp eL42 in catalysis of peptide bond formation, but another protein, namely uL15 is shown to contribute to catalysis along with eL42 at the PTC. In conclusion, when bound to rps eL42 and uL15, the small-molecule inhibitors such as CHX are capable of inhibiting the human ribosomes of the SARS-CoV-2 infected cells, in order to prevent them from synthesizing the viral proteins and enzymes. This would result in antiviral activities corresponding to the blockade of the replication of the viral RNA and the assembly of viral particles. Altogether, the data of the present and previous reports from our group indicate that the statement “no antibiotic for viral infections!” is not relevant for all the clinically approved classes of antibiotics, because selective antibiotics such as the macrolides, the tetracyclines or the glutarimide antibiotics are capable of exhibiting antiviral activities through specific interactions with the human 80S ribosomes of infected host cells.
CONCLUSION
The following concluding remarks can be drawn from the present report: (i) the discovery (if any) of clinically approved vaccines against SARS-CoV-2 will not make it optional to search for specific and effective therapeutic drugs for COVID-19. Therefore, in addition to designing and developing vaccines for SARS-CoV-2, the development of new drugs is also needed; (ii) the majority of scientists, physicians, and healthcare professionals were trained with the paradigm: “antibiotics are for bacteria only!”, because they misunderstand the definition of the ribosome targeting antibiotics and of their targets in the protein translation machinery of the living cells; (iii) after having defined more precisely the ribosome targeting antibiotics in general, and the eukaryote specific glutarimide antibiotics, of which cycloheximide (CHX) is the best known member, in particular, we have broadened the scope of antibiotics and provided evidence that some classes of antibiotics could offer excellent means to counteract viral infections via specific mechanisms exposed in this paper; (iv) in addition, we propose that the large subunit ribosomal protein eL42 would represent a novel therapeutic target for the glutarimide antibiotics and their analogues in SARS-CoV-2-infected host cells; (v) the analogues studied in this report are the thiosemicarbazones that had been previously synthesized in our group by using the structure based drug design approach; (vi) using molecular modeling and docking studies, we have demonstrated that the thiosemicarbazones have potential as therapeutics for COVID-19; (vii) we have compared the binding sites for CHX and its analogue sparsomycin (SPS) on the evolutionarily conserved E.coli bL12 and human eL42, and found that they bind to the conserved catalytic Lys-65 and Lys-53 residues of bL12 and eL42, respectively; (viii) finally, we conclude that the statement “no antibiotic for viral infections!” is not relevant for all the clinically approved classes of antibiotics, because selective antibiotics such as the macrolides, the tetracyclines or the glutarimide antibiotics (or their thiosemicarbazone analogues) are capable of exhibiting antiviral activities through specific interactions with the human 80S ribosomes of infected host cells.
LIST OF ABBREVIATIONS
| rp | = ribosomal protein |
| eL42 | = eukaryal or archaeal large subunit ribosomal protein |
| bL12 | = large subunit ribosomal protein from the bacterial 70S ribosomes |
| PTC | = Peptidyl Transferase Center |
| RdRp | = RNA-dependent RNA polymerase |
| CHX | = Cycloheximide |
| LTM | = Lactimidomycin |
| SPS | = Sparsomycin |
| PLT | = Phyllanthoside |
| SAR | = Structure–Activity Relationship |
| tRNAox | = tRNA dialdehyde derivative |
| TSCs | = Thiosemicarbazones. |
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
HUMAN AND ANIMAL RIGHTS
Not applicable.
CONSENT FOR PUBLICATION
Not applicable.
AVAILABILITY OF DATA AND MATERIALS
Not applicable.
FUNDING
None.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
We are grateful to Drs. Horrhus Hounguè and Soria Baouz-Drahy for fruitful discussions. We gratefully thank Profs. Jean-Marie Schmitter, Pierre Sarthou, Jean Cognet, and Drs. Gérard Keith, Margaret Ahmad and Nathalie Jourdan for their constant interest for this work, and for fruitful discussions.

