All published articles of this journal are available on ScienceDirect.

Nitric Oxide Balance in Health and Diseases: Implications for New Treatment Strategies
Authors Info & Affiliations
Abstract
Nitric Oxide (NO) is an essential signaling molecule with diverse physiological functions in humans. The steady-state concentration and site of production of nitric oxide determine its effects in biological systems. The human cells are exposed to both beneficial and harmful effects of NO. These dual effects of NO could depend on its local concentration in the cells. Additionally, the rate of synthesis, translocation, direct interaction with other molecules, and signals contribute to the biochemical and physiological effects of NO. In this review, the biochemical and physiological role of NO, particularly in health and disease as touching on cell signaling, oxidative stress, immunity, as well as cardiovascular protection amongst others, is focused on. Therefore, this review objectively discusses the dual functionality of NO in living cells.
1. INTRODUCTION
Nitric Oxide (NO) was discovered as a colorless and toxic gas in 1772 by Joseph Priestly. He named it “nitrous air”. Some laboratories conducted research on NO in the late 1970s with no knowledge that they were working on NO molecule. Initially, Gruetter et al. [1] reported that NO induced relaxation of the smooth muscles in the inner lining of the blood vessels, but the mechanism was not established [1]. Another experiment by Furchgott and Zawadzki, after a year, showed that a chemical released by the endothelial cells called Endothelium-Derived Relaxing Factor (EDRF) was triggered by acetylcholine [2]. Subsequently, EDRF, which affects the smooth muscles by initiating the muscle relaxation and vasodilation was discovered to be the NO molecule [3, 4].
Although, NO was discovered in the 18th century, it was not until 1987 that its mechanism of action was known, when Katsuki et al. [5] demonstrated that the NO effects on smooth muscles relaxation was through stimulation of a soluble Guanylyl Cyclase (sGC). Another report by Palmer et al. [6] revealed that NO is catalyzed by NO synthase to produce L-arginine. The NO molecule made headlines in 1992 when it was announced on the cover of Science magazine as “the molecule of the year” [7]. Further events include the introduction of Sildenafil (Viagra®), which affects vasodilation through NO by activation of sGC, by the pharmaceutical company Pfizer to the market and the Nobel Prize award to scientists named Murad, Furchgott, and Ignarro, who unraveled the effects of NO in their studies [8] leading to the worldwide recognition of NO molecule. Further research has been able to show that NO is involved in different physiological processes and pathogenesis of several diseases. For example, the NO was introduced as a signaling molecule in osteoclasts in the late 1990s and in the orthodontic tooth movement in 2002 [8].
As previously reported by Jeandroz et al. [9], the NO is catalyzed by nitric oxide synthase (NOS) enzymes utilizing L-arginine and oxygen. The NO is highly unstable with a short half-life (6¬10 seconds). The NO binds to oxygen, producing NO stable end-products such as nitrate (NO3) and nitrite (NO2) (Fig. 1). It has been demonstrated by Bryan [10], that NOS exists in three isoforms: (1) Neuronal NOS (nNOS or NOS-1), (2) Inducible NOS (iNOS or NOS-2) and (3) Endothelial NOS (eNOS or NOS-3). There are constitutive NOS (cNOS) composed of eNOS and nNOS because they are constitutively expressed in the cells [11].
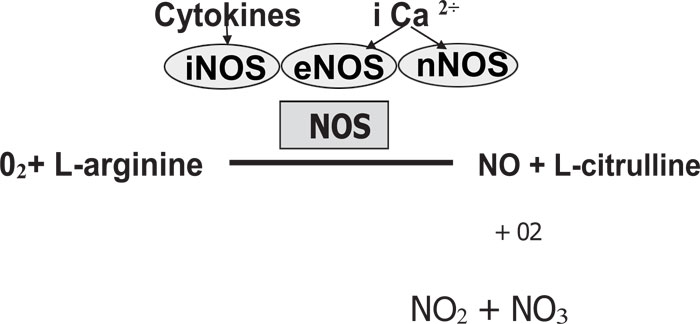
2. CLASSIFICATION OF NITRIC OXIDE SYNTHASE
The different families of NOS are encoded by separate genes [12, 13]. There are different isoforms of NOS classified as either constitutive (cNOS) or inducible (iNOS). nNOS and eNOS are constitutive while iNOS is inducible [14, 15]. Pathogens like Staphylococcus aureus and Bacillus anthraces have demonstrated the activity of NOS [16] and protection against diverse antibiotics, host immune response, and oxidative stress is shown in bacterial NOS (bNOS) [16].
2.1. Neuronal Nitric Oxide Synthase (nNOS)
Neuronal NOS (nNOS) is a constitutive NOS, which produces NO in the nervous system and certain epithelial cells. Neuronal NOS is also associated with plasma membranes and plays a vital role in cell communication. The nNOS activities are regulated by the action of Ca2+ and calmodulin [13], and carried out through vasodilation via peripheral nitrergic nerves, central regulation of blood pressure, smooth muscle relaxation and long-term regulation of synaptic transmission. It also applies to neuronal death of cerebrovascular stroke [17].
2.2. Endothelial Nitric Oxide Synthase (eNOS)
Endothelial NOS (eNOS also called NOS-3) is a constitutive NOS, which regulates vascular function and generates NO in blood vessels [18]. eNOS is Ca2+-dependent NOS, it is also associated with plasma membranes and provides basal NO release.
The three NOS isoforms function as a homodimer during activation, they share a calmodulin-binding domain linked to a heme prosthetic group with an amino-terminal oxygenase domain and also share a cytochrome P450 reductase homologous to the carboxyl-terminal reductase domain [19]. Calmodulin-binding with the amino-terminal oxygenase domain acts as a “molecular switch” to support electron flow in the reductase domain from flavin prosthetic groups to heme. This enables the reaction of L-arginine and oxygen (O2) to produce NO and L-citrulline. The efficient generation of NO required contains a tetrahydrobiopterin (BH4) prosthetic group present in the oxygenase domain of each NOS isoforms. BH4 is recycled by dihydrobiopterin reductase and serves as a source of reducing equivalents. By a single electron donation, BH4 activates heme-bound O2, which can be reobtained to aid the NO release [18].
2.3. Inducible Nitric Oxide Synthase (iNOS)
iNOS is an inducible, calcium-insensitive NOS, possibly as a result of its tight non-covalent interaction with Ca2+ and calmodulin. From a functional perspective, iNOS induction occurs mostly in an oxidative environment, and there is a fast reaction between high NO levels with superoxide leading to the formation of peroxynitrite which then causes cellular toxicity [20]. These properties of iNOS make it essential in host immunity, anti-tumor, and anti-microbial activities [21].
3. NITRIC OXIDE SYNTHASE: REGULATION AND STRUCTURE
NO cannot be stored in the cells as it could be found in other signaling molecules. NOS expression is tightly regulated and thus, controls the amount of NO produced [22]. NOS are well-regulated enzymes in biology. The isoforms of NOS are regulated differently, but showed similarity in structure and are also homodimeric with a linker segment and two oxygenase and reductase domains. There are different binding sites on the isoforms L-arginine, calmodulin, tetrahydrobiopterin, flavin mononucleotide, and flavin adenine dinucleotide [23]. All NOS isoforms are biochemically activated when bound to calmodulin [24]. The constitutive isoforms are mainly regulated by the concentration of intracellular calcium. Increased calcium levels enhance NOS binding to calmodulin and lead to isoform activation [22].
For an effective and sensitive mechanism, calcium channels are located in proximity to NOS enzymes. The regulation of NO production occurs at both upstream and downstream controlling pathways. Protein interactions and modifications of post-translational level are controlling mechanisms that mediate the upstream controlling pathways. The downstream control occurs through NOS localization in proximity to NO target molecules. The diffusion of NO to its target cells is because it is a free radical that reduces its toxic effects in the surrounding environment [25].
iNOS is regulated differently compared to the regulation of nNOS and eNOS. iNOS activities are not affected by the levels of intracellular calcium, but it binds to calmodulin just like other NOS. However, in low levels of intracellular calcium, iNOS still bind to calmodulin, which means its activation may be independent of calcium concentration [26].
4. SYNTHESIS OF NITRIC OXIDE
Nitric oxide synthase catalyzes nitric oxide synthesis and three NOS isoforms with different function; inducible (iNOS), endothelial (eNOS) and neuronal (nNOS) (Fig. 2). The endothelial and neuronal isoforms are characterized by low NO production as a signaling molecule and are also calcium-dependent. The inducible isoform is calcium-independent and can be cytotoxic due to large amounts of NO gas produced. The NOS reacts with the guanidine group of L-arginine through oxidation, thereby losing five electrons to form L-citrulline and NO. The NADPH oxidation and reduction of molecular oxygen are involved in the reaction. Adjacent to the L-arginine specific binding site, the catalytic transformation occurs [27].
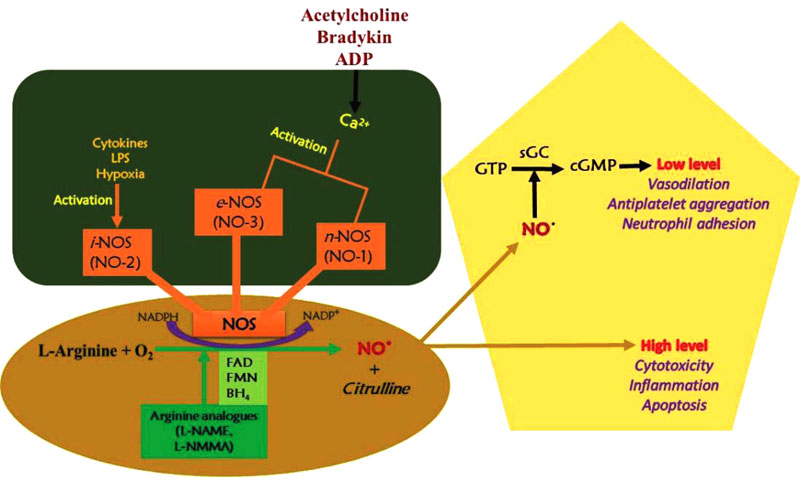
The neuronal NOS acts as a transmitter and is found in different nerves in the nervous system, like non-cholinergic (NANO) and non-adrenergic autonomic nerves [28]. In blood vessels, constitutive nitric oxide synthase (cNOS) activities produce NO in a calcium- and calmodulin-dependent reaction. Stimulation of cNOS occurs in two basic pathways, which involve calcium ions release from sub-sarcolemma storage sites.
In physiological conditions, iNOS activity is minimal and can be stimulated during inflammation and cytokines, such as tumor necrosis factor (TNF) and interleukins. About a 1,000-fold of NO is produced by iNOS during inflammation than that produced by cNOS. The NO production by endothelial nitric oxide synthase is stimulated by cytokines, acetylcholine, platelet-derived factors, and shear stress [29].
4.1. Intracellular Mechanisms
NO has a very short life after its production before it becomes oxidized to one of its more stable end-products. There is a high affinity between superoxide anion and NO as a result of an unpaired electron in their outer shell, making them highly reactive and thus, reducing NO bioavailability. In most cells, NO binds to the heme moiety of hemoglobin and guanylyl cyclase [30]. Therefore, there is rapid diffusion into the blood when NO is formed and then binds to hemoglobin. Activation of guanylyl cyclase occurs when NO binds to guanylyl cyclase and diffuses into the smooth muscle cells [30].
The synthesis of NO and its subsequent effects are dependent on the types of cells that produce NO and also conditions experienced by the organism during the production periods. For instance, the production of NO by vascular endothelial cells in small amounts is continuous and this contributes to the maintenance of blood homeostasis and normal blood pressure [31]. During septic shock, the expression of NOS-2 induces the vascular endothelial cells to produce high NO concentrations, a process associated with persistent hypotension, decompensation, and vasoplegia [32, 33].
4.2. Mechanism of Action
Nitric oxide provides several mechanisms that affect cell biology, for instance, guanylate cyclase activation, nitrosylation of protein sulfhydryl group, ADP ribosylation of proteins, iron-containing proteins oxidation, such as aconitase and ribonucleotide reductase, and iron regulatory factor activation [34]. In peripheral blood mononuclear cells, NO activates a vital transcription factor in the iNOS gene expression named nuclear factor kappa light chain enhancer of activated B cells (NF-KB) as a result of inflammation. NO acts through soluble guanylate cyclase stimulation to subsequently form cyclic GMP. The cGMP activates protein kinase G to inactivate myosin light-chain kinase. This eventually dephosphorylates myosin light chain and results in the relaxation of the smooth muscles [35].
4.3. Physiological and Cellular Action
NO is important in regulating and mediating several processes in the immune, cardiovascular, and nervous systems, such as neurotransmission and relaxation of smooth muscles as a result of vasodilation and eventually increasing blood flow [36]. In the pathophysiological state, NO has been implicated in conditions, such as septic shock, stroke, neurodegenerative diseases, and hypertension [37].
Guanylate cyclase activation by NO causes the relaxation of smooth muscle by:
- Decreasing intracellular calcium concentrations by increasing the levels of intracellular cGMP
- K+ channels activation resulting in relaxation and hyperpolarization
- cGMP-dependent protein kinase stimulation stimulates myosin light chain phosphatase, thereby leading to smooth muscle relaxation.
4.4. Vasodilation
NO, induced by several factors, mediates vasodilation in blood vessels. Its synthesis by eNOS stimulates smooth muscles relaxation through the phosphorylation of several proteins. The vasodilatory action is vital in blood flow regulation, renal control of extracellular fluid homeostasis, and maintenance of blood pressure [13]. After synthesis, due to its highly reactive nature, it diffuses into the smooth muscle cells and interacts with soluble guanylate cyclase. The soluble guanylate cyclase is stimulated by NO to produce cGMP from GTP [38]. The soluble cGMP activates cyclic nucleotide-dependent protein kinase G (PKG or cGKI) and results in the relaxation of smooth muscles [39].
Additionally, the vasodilatory effect of NO function is in erection development and maintenance. The blood vessels vasodilation results in increased blood flowing into the corpus cavernosum, which leads to erection [40]. Sildenafil (Viagra) follows a similar mechanism by inhibiting phosphodiesterase-5 (PDE5), which lowers the concentration of cGMP by reconverting to GMP.
4.5. Immune System
In the immune system, the macrophages produce NO using iNOS so as to eliminate the invading bacteria [41]. For example, fulminant infection (sepsis) causes excess NO production by macrophages, and could probably cause hypotension in sepsis as a result of vasodilatation. The iNOS, when expressed, generates cytotoxic levels of NO [42].
4.6. Inflammatory Action
During inflammatory reactions in humans, there is a high level of NO produced and iNOS is principally responsible for this process. At low concentration, NO is a pro-inflammatory agent that induces vasodilatation and recruit neutrophils, while high concentrations of NO downregulate adhesion molecules, suppresses activation, and induce apoptosis of inflammatory cells [43, 44]. During inflammatory reactions, there is an increased expression of iNOS by pro-inflammatory cytokines in cells like neutrophil granulocytes and monocyte/ macrophages. However, endotoxin induces expression strongly in bacterial infection. As a result, there is a high production of NO, which exceeds the physiological NO by up to 1000-fold. As an inflammatory mediator, the overproduction can result in tissue destruction, such as in inflammatory autoimmune diseases. Thus, NO can process pro- or anti-inflammatory effects depending on the concentration [45]. Several reports have shown the relationship between inflammation in the brain and the pathogenesis of several neurodegenerative disorders, such as Alzheimer’s diseases, dementia, Parkinson’s disease, amyotrophic lateral sclerosis, AIDS, and multiple sclerosis. The brain inflammation is mainly caused by the activation of glial cells, which stimulate the production of several pro-inflammatory cytokines. Neurodegenaration has been attributed to the excessive production of NO due to NOS induction in activated glia. Increased NO production has also been linked with chronic inflammatory bowel diseases, arthritis, and inflammatory diseases of the respiratory tract [45, 46].
5. NITRIC OXIDE IN THE CELLULAR ENVIRONMENT
5.1. Lungs
The lung is composed of numerous cell types, such as macrophages, epithelial cells, endothelial cells, vascular smooth muscle cells, bronchial smooth muscle cells, neurons, and pneumocytes [47]. Each cell type produces NO through one or more isoforms of NOS [48]. Blood vessels irrigate all the components in the lungs, while essential blood vessel components named endothelial cells, express NOS-3 [49]. The NO in the lungs is involved in the production of mucin by the bronchial epithelial cells, the participation of macrophages in phagocytosis, bronchodilation at the inhibitory non-adrenergic /non-cholinergic nerve terminals, and vasodilation at the endothelium [50]. These functions are controlled and coordinated to aid adequate blood flow as well as in allergen- or immunogen-induced inflammation [50].
The NO and reactive species, such as RNS, are essential in different inflammatory pulmonary diseases [51]. For example, the exhaled air of asthma patients showed increased NO levels and it is consistent with elevated gene expression of NOS -2 [52, 53]. A pathological lung condition called chronic obstructive pulmonary disease is associated with pulmonary hypertension [54], the NO level in patients’ exhaled air was demonstrated to be elevated two-fold higher than normal [55]. As in the case of asthma, increased oxidative stress in the lung has been associated with COPD [56].
5.2. Liver
All the NOS isoforms are produced in hepatocytes. Different conditions, however, can modulate the synthesis and effects of NO, such as contact with metabolic by-products, oxygen availability, and other various cellular interactions [57]. Although, it has been shown that the hepatocyte gene profile and metabolic functions are dependent on its location [58]. Hence, it is reasonable to claim that NOS isoforms expression and the NO effects are dependent on enzyme compartmentalization, cell localization in the organ, and also the developmental timing of enzyme expression. An example of changes from different NOS compartmentalization is that a change in the hepatic blood circulation during endotoxic shock has been found to be anti-parallel to those observed in the lungs [47].
5.3. Skeletal Muscle
Nitric oxide carries out a vital role in the contraction of the skeletal muscle [59]. NOS-2 and NOS-3 found in the skeletal muscle are largely located in the sarcoplasm, while NOS-1 is found only in sarcolemma [60], anchored by two proteins named syntrophin and dystrophin, that forms the dystrophin protein complex [61]. For instance, the gene encoding dystrophin is mutated in Duchenne Muscular Dystrophy (DMD), and as a consequence, NOS-1 remains localized in the sarcoplasm even as NOS-1 mis-localization has shown to contribute towards muscular ischemia [62].
6. NITRIC OXIDE IN VARIOUS CELLULAR CONDITIONS
6.1. Effects of Nitric Oxide (NO) on the Cardiovascular System
NO potentially influences vascular homeostasis through different means, such as vasomotion modulation, platelet, and monocyte adhesion to the endothelium, production of endothelin, inhibition of smooth muscle cell proliferation, aggregation of platelet, expression of adhesion molecules and oxidation of LDL. Decreased bioavailability of NO, as a result of defects in function, are caused by decreased activities of antioxidant enzyme/increased oxidative or nitrative stress and are responsible for diseases, such as vascular restenosis, septic shock, sickle cell diseases, immune reaction, coronary artery disease, inflammation, peripheral vascular disease, diabetes, hypercholesterolemia, atherosclerosis, migraine, meningitis, hypertension, and stroke [63, 64].
In normal vascular biology, there is a need for interaction between endothelial cells and monocytes, but lengthy interactions are implicated in the pathogenesis of vascular diseases [65]. The interaction lowers the release of biologically active NO and develops into atherosclerotic lesions. The NO is an anti-proliferative, anti-atherogenic, and anti-thrombotic factor [66].
6.2. Nitric Oxide in Infection
In understanding the pathogenesis of infection, several studies have been carried out over the last two decades showing the importance of enzymatically produced NO in diverse physiological processes. The morbidity of the host during an infection is contributed by NO by acting as a myocardial depressant, vasodilator, and cytotoxic mediator. NO possesses immunoregulatory, cytoprotective, microvascular, and anti-microbial properties that have a beneficial role in the infected host [67].
Evidences implicate the generation of NO as an effective host response to infection. The generation of NO by the iNOS isoform (NOS-2) is stimulated by pro-inflammatory cytokines. Infection in humans and experimental animals significantly increases in systemic NO production, when determined by measurement of NO oxidation end-products (nitrite and nitrate) in plasma and urine [68]. In animal models, infections, such as leishmaniasis and toxoplasmosis, as well as tuberculosis in humans, have shown to have elevated expression of NOS-2 and NO production [69, 70]. Possibly, there is a correlation between NO production and the host ability to contain microbial proliferation, due to dramatic increases in microbial burden as a result of abrogation of inducible NOS activity [71, 72]. In vitro studies revealed that NO-donor compounds could kill or inhibit microbial activities when administered directly [69].
The NO scavengers’ abundance outside the phagosomal compartment helps the host defenses against organisms, such as Salmonella, Mycobacteria, and Leishmania. NO scavenging by hemoglobin in an experimental system can physiologically antagonize anti-microbial action of NO against bacteria and trypanosomes, which is relevant in bloodstream infection or haemorrhage [73]. Several pathogens, such as Toxoplasma gondii, Mycobacterium tuberculosis, and Leishmania spp. promote persistent asymptomatic latent infection. However, reactivation of some organisms has been induced in experimental models, such as Leishmania major, T. gondii, M. Tuberculosis, or Epstein -Barr virus, due to NO synthesis inhibition [70], indicating the major role of NO in latent infections.
6.3. Sepsis
Under normal physiological conditions, NO derived from eNOS present in the smooth muscle stimulates soluble guanylate cyclase, which enhances intracellular cGMP and results in vasodilation. In sepsis, iNOS derived NO mimics and amplified the process to cause chronic vasodilation and damage other pathways. Thus, iNOS becomes an important mediator of shock and organ damages in sepsis. Animal models affirm widespread NO production and iNOS expression in response to inflammatory cytokines, endotoxin, or Gram negative bacteria, causing organ failure, hypotension, and death [74].
6.4. Hypertension
Decreased bioavailability of NO is an essential mechanism in the pathogenesis of hypertension. A report has revealed that angiotensin II- (Ang II-) induced hypertensive rats showed eNOS phosphorylation at threonine 495 residue [75]. Several investigations have observed in salt-induced hypertension rats that there is high BH4 oxidation as a result of activation of the p47phox subunit of NADPH oxidase [76, 77]. Another report suppresses hypertension in spontaneously hypertensive rats after BH4 was orally administered thanks to the O2− accumulation and reduction of ONOO− [78].
6.5. Diabetes
Diabetes involves a reduced synthesis of NO production. Reports have shown that eNOS activity was reduced by increased phosphorylation at threonine 495 via PKC in the obese mouse model [79]. Alp et al. showed, in diabetic hypertensive rats, decreased synthesis of NO and low BH4 levels [80]. Heitzer and colleagues demonstrated that supplementation of BH4 improved endothelium-dependent vasodilation in patients with type II diabetes but not in control subjects. Such beneficial effect was completely blocked by N(G)-monomethyl-L-arginine, a well-known inhibitor of NOS, suggesting that it was dependent on the NO production increase [81].
CONCLUSION
Besides mediating normal functions, such as smooth muscle relaxation causing the artery vasodilation and increased blood flow; macrophage mediated cytotoxicity against microbes and tumor cells and neurotransmission, NO has also been implicated in several essential disease conditions. Interestingly, some disease processes are linked with excess production of NO, while other conditions, such as achalasia, are connected with low levels of NO. Therefore, the view into the mechanistic biochemistry of NO in health and disease is a requirement for the NO-related newer and improved therapies.
CONSENT FOR PUBLICATION
Not applicable.
FUNDING
None.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
Declared none.

