All published articles of this journal are available on ScienceDirect.

Purification, Characterization and Comparison between Two New L-asparaginases from Bacillus PG03 and Bacillus PG04
Authors Info & Affiliations
Abstract
Background:
L-asparaginase has been used as a chemotherapeutic agent in treatment of lymphoblastic leukemia. In the present investigation, Bacillus sp. PG03 and Bacillus sp. PG04 were studied.
Methods:
L- asparaginases were produced using different culture media and were purified using ion exchange chromatography.
Results:
Maximum productivity was obtained when asparagine was used as the nitrogen source at pH 7 and 48 h after cultivation. New intracellular L-asparaginases showed an apparent molecular weight of 25 kDa and 30 kDa by SDS-PAGE respectively. These enzymes were active in a wide pH range (3-9) with maximum activity at pH 6 for Bacillus PG03 and pH 7 for Bacillus PG04 L-asparaginase. Bacillus PG03 enzyme was optimally active at 37 ˚C and Bacillus PG04 maximum activity was observed at 40˚C. Kinetic parameters km and Vmax of both enzymes were studied using L-asparagine as the substrate. Thermal inactivation studies of Bacillus PG03 and Bacillus PG04 L-asparaginase exhibited t1/2 of 69.3 min and 34.6 min in 37 ˚C respectively. Also T50 and ∆G of inactivation were measured for both enzymes.
Conclusion:
The results revealed that both enzymes had appropriate characteristics and thus could be a potential candidate for medical applications.
INTRODUCTION
L-Asparaginase (EC.3.5.1.1; L- asparagine amidohydrolase) enzyme has been extensively studied in mammals and microorganisms because of its potential antineoplastic activity [1]. L-asparaginase acts by catalyzing the conversion of amino acid L-asparagine into aspartic acid and ammonia. Hence, this enzyme is very useful in maintaining the level of amino acid within the cells [2].The use of this enzyme in anti-cancer therapy is based on its L-asparagine cleavage activity of the enzyme. L-asparagine is an essential amino acid for lymphoblast s’ growth. The enzyme acts by reducing the accessible amino acid, thus depriving tumor cells which are unable themselves to synthesize this amino acid whereas normal cells are able to make their own asparagine [3, 4]. L-asparagine deficiency rapidly impairs the protein synthesis and leads to an impairment of cellular function, resulting in cell death [5]. The enzyme can also be used to reduce the formation of acrylamide in fried and oven-cooked foods especially in potato chips. Since acrylamide formation in heated foods is mainly due to the reaction of free asparagine and reducing sugars, deamination of asparagine prevents acrylamide formation [6, 7]. L-asparaginase enzyme has been isolated from various sources [8-10]. Microbial enzymes are preferred over plant or animal sources due to their economic production, consistency, ease of process modification, optimization and purification [11-15]. These enzymes are also more stable than the corresponding plant or animal derived enzymes. Hence, microbial sources are best for the bulk production of L-asparaginase for clinical uses [16]. Although L-asparaginase is produced by various microorganisms [17], enzymes isolated from Escherichia coli [18] and Erwinia carotouora [19] are now being used for the treatment of acute lymphoblastic leukemia. The main restriction of these enzymes in the therapeutic field is several types of side reactions, from mild allergies to dangerous anaphylactic shocks [20]. Also L-glutaminase activity is a reason for enzyme toxicity. Therefore, there is a need to find novel enzymes with new characteristics, different serological properties and higher therapeutic activity. In our previous study, we isolated and identified two L-asparaginase producing bacteria, Bacillus PG03 (NCBI accession number: KF150763) and Bacillus PG04 (NCBI accession number: KF150760) from the Persian Gulf [21]. In the present investigation, we have studied purification and partial characterization of L-asparaginase enzyme produced by these two bacteria. The effect of limited nitrogen sources and pH on the production of L-asparaginase has been investigated. Kinetic and thermal parameters including t1/2, T50 and ΔG* of the enzymes have been studied. Also optimum pH and temperature of the enzymatic activity have been evaluated. Since L-asparaginase derived from E.coli is used as chemotherapeutic agent, the properties of this enzyme were also compared with the enzymes produced by Bacillus PG03 and Bacillus PG04.
MATERIALS AND METHODS
Chemicals
L-asparagine, L-glutamine, Trichloro acetic acid and Tris were purchased from Merck (Darmstadt, Germany). DEAE-Sepharose was provided by Pharmacia (Uppsala, Sweden). L-asparaginase derived from E.coli was a gift from cancer research institute, Iran. All other chemicals were from Sigma (St. Louis, MO, USA) and were of analytical grade.
Microorganism Media and Growth Conditions
Two new bacterial strains, Bacillus PG-03 and Bacillus PG-04 were previously isolated from the Persian Gulf sediments and identified. Colonies were sub-cultured on to nutrient agar slants and kept in 20% glycerol at -20 ˚C as stock culture. The primary inoculum was grown overnight in 10 ml nutrient broth medium and was inoculated in 100 ml of M9 broth, consisting of (g/L): maltose 20% as a sole carbon source; Na2HPO4 6.0; KH2PO4 3; NaCl 0.5; CaCl2 0.011; MgSO4.7H2O.7H2O 0.12; pH 7.0 and incubated at 37 ˚C with shaking at 200 rpm for 24 hours. The cell mass concentration was determined by measuring the optical density of the culture at 600 nm.
Effect of Nitrogen Sources and pH
To study the effect of different nitrogen sources on L-asparaginase activity, alternative nitrogen sources consisting of L-asparagine, arginine and NaNO3+(NH4)2SO4 (1% w/v) were used. pH 7 and pH 8 were tested for maximum enzyme production.
Enzyme Extraction and Purification
For extraction of the intracellular enzyme, the sonication method was used. The biomass was separated out by centrifugation (8000 rpm, 10 min and 4 ˚C); the pellet was resuspended in 3mL 50mM Tris buffer pH 9 containing 10% (v/v) glycerol. The cells were disrupted by sonication and then centrifuged at 13,000 rpm and at 4 ˚C for 15 min. The supernatant used as the crude extract. The crude enzyme solution was applied to DEAE-Sepharose column (10mm×100 mm) that was pre-equilibrated with a 50mM Tris-HCl buffer, pH 9.0. The protein was eluted with the NaCl gradient (0.0-0.7 M) and 50 mM Tris-HCl buffer, pH 8. The active fractions were collected, dialyzed and concentrated. This preparation was used in the subsequent steps.
Molecular Weight Determination
SDS-PAGE was carried out using a 12% polyacrylamide gel by the method of Laemmli [22]. A high molecular weight marker (Fermentas) was used. Protein bands were detected by Coomassie Brilliant Blue R250 and destained with a solution of methanol, acetic acid and water in the ratio of 4:1:5.
Enzyme Activity, Protein Concentration and Biochemical Characterization
L-asparaginase activity was measured by the known method of Imada et al. [23]. The samples were mixed with 40 mM L-asparagine in 50 mM Tris-HCl buffer, pH 7.2. 200 µL of assay mixture were incubated for 15 min at 37 °C for enzymatic reaction. Reaction was stopped by the addition of 250 μL of 1.5 M Trichloro acetic acid (TCA) and samples were centrifuged before the addition of Nessler’s reagent to measure the released ammonia after L-asparagine hydrolysis. Absorbance was measured at 450 nm. One unit of L-asparaginase activity was defined as the amount of enzyme that released 1 μmol of ammonia (with ammonium sulfate as the standard) per minute under the assay conditions specified. L- glutaminase activity was determined as that of L-asparaginase using the method of Mashburn and Wriston [18]. Protein concentration was also measured according to the method of Bradford using bovine serum albumin as the standard [24]. The pH profile and pH optima were determined at room temperature in various pH using 50 mM phosphate buffer. The activity of purified enzyme was determined at several temperatures (from 25 to 50 ˚C) in 20 mM Tris-HCl buffer, pH 7.2.
Kinetic Parameters
In order to determine kinetic constants for L-asparaginase, measurements were carried out over different substrate concentrations including 10, 20, 30, 40 and 80 mM and the final concentration of the enzyme was 0.5 mg/mL. Different blanks were used for each L-asparagine concentration. Kinetic parameters, Km and Vmax for L-asparaginase enzymes were determined from a series of initial rates. Experimental data were analyzed graphically and numerically using the Lineweaver–Burk equation.
Thermal Stability Assay
Half-lives and T50 were determined at two temperatures (37, 40˚C). For half-life measurement, 1 mg/mL enzyme solution in 20 mM Tris–HCl buffer (pH 7.2) was used. 10 μL aliquots were removed at various time intervals, chilled on ice for 60 min and diluted into 20 μL of an assay solution (L-asparagine 40 mM) for the measurement of residual activity at 37˚C for 15 min. Plots of the log of residual activity versus time were linear, indicating a first-order decay. T50 is the temperature of incubation at which 50% of the initial enzyme activity is lost during 30 min incubation [25]. The ΔG* parameter of the L-asparaginase enzyme was determined as follows [26]:
ΔG * = RTln (kBT/h) - RTlnk
RESULTS AND DISCUSSION
Cultivation Medium and Culture Conditions
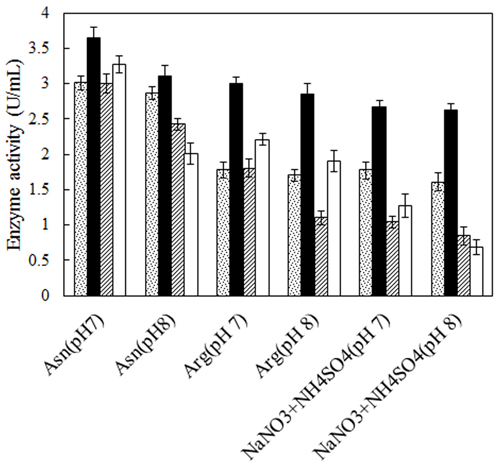
No defined medium has been established for the optimum production of L-asparaginase from different microbial sources. Each organism has its own special conditions for maximum enzyme production. The prerequisite for obtaining L-asparaginase from bacteria is to grow them in a medium optimized with various nitrogen sources [27, 28]. Therefore, two bacterial strains, Bacillus PG03 and Bacillus PG04 were screened for the L-asparaginase production at various nitrogen sources and pH. Three different nitrogen sources including L-asparagine, L-arginine and NaNO3+(NH4)2SO4 were separately added to the basal medium containing maltose as carbon source, pH 7 and pH 8. The results revealed that for both bacterial strains, maximum L-asparaginase production obtained in the presence of L-asparagine as the nitrogen source at pH 7. The cells were cultivated in each media for 24 h and 48 h. The enzyme production was higher after 48 h for both strains (Fig. 1). Previous studies have shown that a nitrogen source is the limiting factor and plays key role in the L-asparaginase production [29]. It seems that L-asparagine might be an inducer for L-asparaginase production and hence the production of the enzyme is more feasible at the presence of this nitrogen source compared to L-arginine and NaNO3 + (NH4)2SO4. Maximum L-asparaginase production using L-asparagine as the sole source of nitrogen has been observed in Streptomyces gulbargensis [30], Enterobacter cloacae [31] and Aeromonas sp [32]. On the contrary, Narayana et al. reported yeast extract (2%) as the best nitrogen source for L-asparaginase production by S. albidoflavus [33].
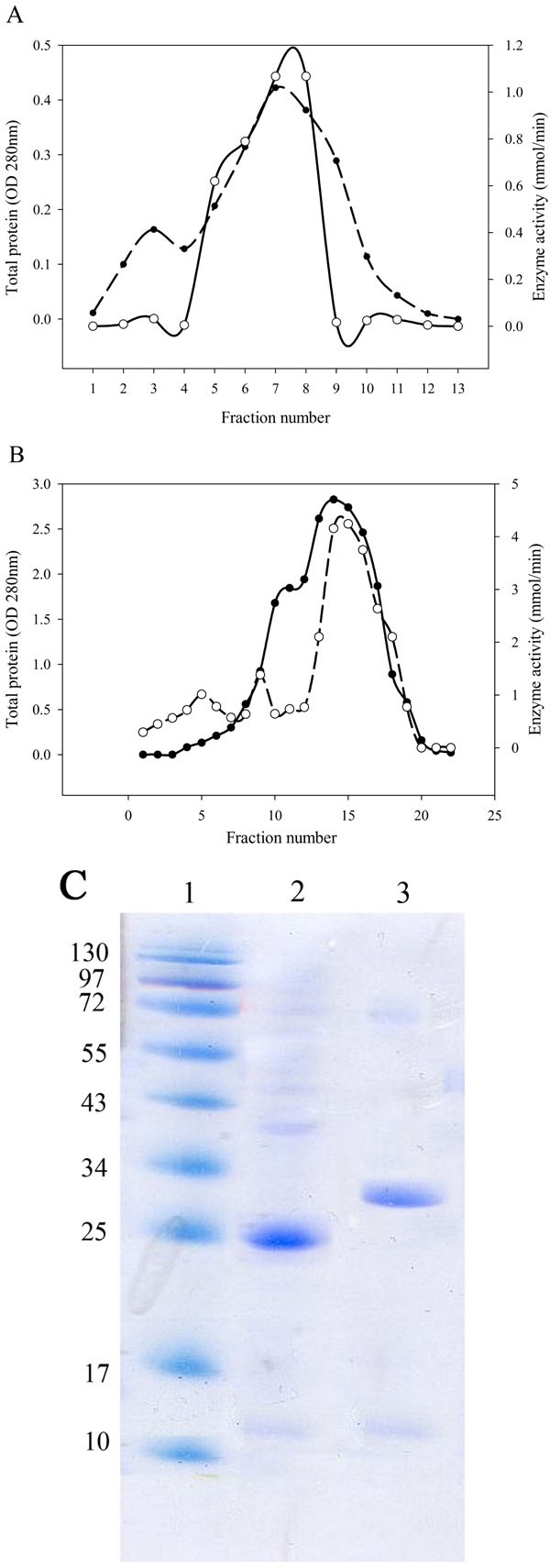
L-asparaginase Purification and Molecular Weight Determination
After bacterial cell sonication, the crude enzyme was applied to anion-exchange chromatography on DEAE-Sepharose. The fractions of the largest peak from the elution profile that had L-asparaginase activity were pooled, dialyzed and concentrated by ultrafiltration (Fig. 2A, B). SDS-PAGE analysis of the purified enzyme estimated that the apparent molecular weight of the purified enzyme was 25 kDa for Bacillus PG03 and and 30 kDa for Bacillus PG04 (Fig. 2C). Since most of the bacterial L-asparaginases exist as a tetramer of identical subunits, then the exact estimation of the molecular weight requires a native PAGE analysis.
Kinetic Parameters and Biochemical Characterization
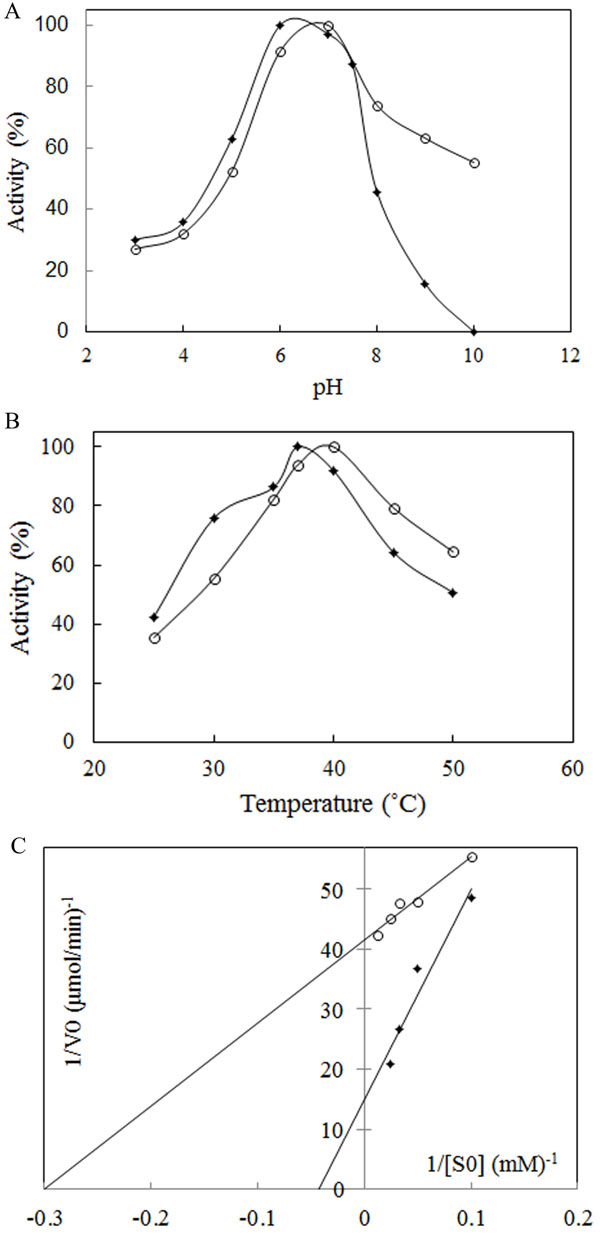
The most important physical factors, which influence the enzymatic reaction rate, are pH and temperature of incubation with the substrate. Each enzyme has a characteristic pH and temperature optima beyond which the reduction in activity is observed. The suitable temperature and pH of the enzyme was studied using a 50 mM phosphate buffer of different pH values, ranging from 3.0 to 10. The results showed that L-asparaginase was active over broad pH ranges from 3 to 8 for Bacillus PG03 L-asparaginase and from 3 to 10 for Bacillus PG04. The enzyme activity gradually increased until pH 6 for Bacillus PG03 L-asparaginase and pH 7 for Bacillus PG04 at which the maximum activity was observed (Fig. 3A). E. coli L-asparaginase was optimally active at pH 6 (Supplementary Data Fig. ( 1A)). These results indicated that the optimum pH and temperature lie near that of the growth condition. Along with E. coli L-asparaginase, enzymes isolated from Erwinia carotouora are now being used for the treatment of acute lymphoblastic leukemia [19]. However, Kamble et al. found that the optimum pH of the enzyme from E. carotouora was 8.6 [34]. Our results revealed that the L-asparaginase produced by Bacillus PG03 and Bacillus PG04 were maximally active around pH 7 which is near to the physiologic pH. Since the physiological pH is one of the perquisites for antitumor activity [35], hence this characteristic makes the enzyme highly attractive for both the basic research studies and the industrial and pharmacological processes. Different optimum pH, from 6 to 9.5 has been reported for enzyme produced by microbial sources [11, 36]. There are very few reports in which the L-asparaginase enzymes showed optimum pH near physiologic pH range [37-39]. The majority of L-asparaginases from Erwinia sp. showed alkaline pH optima (8.0–9.0) whereas the enzyme from E. coli exhibited an acidic pH optimum of 5.0–6.0 [40, 41]. Similarly, the effect of temperature on the activity of L-asparaginase was studied. Maximum activity was obtained at 37˚C for Bacillus PG03 L-asparaginase and 40˚C for Bacillus PG04 and E. coli L-asparaginase (Fig. 3B) and Supplementary Data Fig. ( 1B)). However the enzyme from both bacterial strains was active at a wide range of temperature condition from 25˚C to 50˚C and retained 50% (Bacillus PG03) and 60% (Bacillus PG04) activity in 50˚C. This pattern was similar to the temperature pattern of L-asparaginase derived from E. coli. This property makes the enzyme more suitable for complete elimination of asparagine in patients suffering from leukemia. Mannan et al. [35] also found 37˚C to be the optimum temperature for the enzyme activity. Mesas et al. reported 40˚C as the optimum temperature of the L-asparaginase from Corynebacterium glutamicum [42]. Anyway, a literature review from Zuo et al. and from Batool et al. reported different optimum temperatures from 30˚C to 80˚C for different microbial L-asparaginases [11, 36]. The L-asparaginase enzymes were assayed for amidohydrolase activity. No activity was determined when L-Glutamine was used as substrate. This is consistent with the results of Chityala et al. and Kumar et al. who reported glutaminase free L-asparaginase enzyme [43, 44].The enzymes from Bacillus PG03 and Bacillus PG04 revealed Michaelis-Menten kinetics when L-asparagine was used as a substrate. (Table 1) and Fig. (3C) shows the Km and Vmax values calculated based on the Lineweaver-Burk analysis. Also the kinetic parameters of the E. coli L-asparaginase was studied (Supplementary Data Fig. ( 2A)) Previous studies have reported that L-asparaginases from different microorganisms had different substrate affinities and probably played different physiological roles. Higher and lower Km values have been reported for different L-asparaginase enzymes [8, 45]. E. coli L-asparaginase showed Km of 3.4 mM which is similar to that of Bacillus PG04 and lower than the Km value of Bacillus PG03.

| Enzyme | Kma (mM) | Vmaxa (μmol/min) | pHb | Tcopt (°C) | Tc50 (°C) | td1/2 37°C (min) | td1/240°C (min) |
∆G*d (kcal/mol) |
|---|---|---|---|---|---|---|---|---|
| Bacillus PG03 | 23.8 | 0.067 | 6 | 37 | 49 | 69.3 | 40.8 | 5.4 |
| Bacillus PG04 | 3.3 | 0.024 | 7 | 40 | 48 | 34.6 | 23 | 4.97 |
Thermal Stability Properties
The method that has been used to analyze the heat inactivation rate constants of L-asparaginase requires enzyme incubation at desirable temperatures and cooling in ice before measurement of the residual activity [25, 46]. After incubation at 37˚C and 40˚C, the half-lives (t1/2) of Bacillus PG03 and Bacillus PG04 were measured (Table 1). ki (inactivation rate constant) values were estimated from the plots of Log of residual activity versus incubation time. As is shown in Fig. (4A) inactivation process was a first order decay. L-asparaginase from both strains showed higher half-life in 37˚C compared to 40˚C. Bacillus PG03 L-asparaginase showed the highest half -life of more than 1 hour in 37˚C. This makes the enzyme potentially suitable for future therapeutic uses. Bacillus PG04 enzyme also revealed t1/2 of 34.6 min in 37˚C. In 40˚C still both enzymes were active, although the reduction in half- life of the enzymes was clearly observed compared to 37˚C. The interrelationship between conformational stability and enzyme activity suggested that in naturally occurring enzymes the highest stability could be seen at temperatures near that of growth of an organism [28, 47]. E. coli L-asparaginase showed the half-life of more than 6 hours (Supplementary Data Fig. ( 2B). Together these results showed that although the purified enzymes had appropriate stability in 37˚C and 40˚C but when compared to E. coli L-asparaginase, the half- life of the enzyme needs to be more improved by protein engineering methods and hence these enzymes could be regarded as an interesting target for further investigations. Another criterion of thermal stability is T50. As is shown in Fig. (4B) and Table 1, T50 value for Bacillus PG03 is slightly higher than that of Bacillus PG04. Free energy changes of inactivation (ΔG*) was evaluated for both enzymes. L-asparaginase from Bacillus PG03 displayed a higher ΔG* values of inactivation than the enzyme from Bacillus PG04, indicating that the distance between the native state and the transition state has increased in L-asparaginase from Bacillus PG03 and, consequently, a larger input energy is required for inactivation (Table 1). Results of this study suggest that L-asparaginase from Bacillus PG03 is thermally more stable than that of Bacillus PG04.
CONCLUSION
Finally this investigation revealed that the bacterial strains Bacillus PG03 and Bacillus PG04 produce L-asparaginase enzymes which are optimally active at 37˚C and at pH near physiologic range. These two features of the enzymes are very important for the future therapeutic uses. Only the enzyme produced by H. pylori CCUG 17874 showed similar characteristics [37]. The L-asparaginases of these two strains have a broad temperature range of activity. In comparison to Bacillus PG04, the enzyme produced by Bacillus PG03 showed higher half-life at 37˚C, higher T50 and higher ∆G*. However Bacillus PG04 exhibited lower km and thus higher affinity to L-asparagine as substrate. Therefore, L-asparaginase from Bacillus PG03 is thermally more stable and the enzyme from Bacillus PG04 is kinetically more beneficial. In comparison to the L-asparaginase enzyme produced by E. coli, the reported enzymes had better optimum pH and temperature but the half-life of the enzyme does not seem appropriate for therapeutic uses. Hence, thermal characteristics of both enzymes need to be improved by thermal stability improvement investigations. Since patients showing sensitivity to one formulation of L-asparaginase are switched to other sources of the enzymes, then finding of other enzyme sources is in progress [12, 36]. For example, in addition to E.coli and Erwinia asparaginase, a novel recombinant E. coli asparaginase preparation is currently being subjected to clinical evaluation [48]. Due to the importance of our recent findings, further enzymatic studies, structural analysis, determination of encoded gene sequence of these L-asparaginases and sequence comparison of these enzymes with that of E.coli can be carried out in order to understand the mechanism of its activity and to improve kinetic and stability properties of these therapeutic targets by protein engineering methods.
SUPPLEMENTARY MATERIAL
Supplementary material is available on the publishers Website along with the published article.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
The authors express the gratitude to the molecular medicine research center and research council of Hormozgan University of medical sciences, Iran for the financial support.
REFERENCES
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]

