All published articles of this journal are available on ScienceDirect.

Distinct Metal Ion Requirements for the Phosphomonoesterase and Phosphodiesterase Activities of Calf Intestinal Alkaline Phosphatase
Authors Info & Affiliations
Abstract
The roles of Mg2+ and Zn2+ ions in promoting phosphoryl transfer catalysed by alkaline phosphatase are yet to be fully characterised. We investigated the divalent metal ion requirements for the monoesterase and diesterase activities of calf intestinal alkaline phosphatase. The synergistic effect of Mg2+ and Zn2+ in promoting the hydrolysis of para-nitrophenyl phosphate (monoesterase reaction) by alkaline phosphatase is not observed in the hydrolysis of the diesterase substrate, bis-para-nitrophenyl phosphate. Indeed, the diesterase reaction is inhibited by concentrations of Mg2+ that were optimal for the monoesterase reaction. This study reveals that the substrate specificities of alkaline phosphatases and related bimetalloenzymes are subject to regulation by changes in the nature and availability of cofactors, and the different cofactor requirements of the monoesterase and diesterase reactions of mammalian alkaline phosphatases could have significance for the biological functions of the enzymes.
INTRODUCTION
Alkaline phosphatases (APs) are dimeric metalloenzymes (Fig. 1) that catalyse the hydrolytic transfer of phosphate to water or its transphosphorylation to amino alcohols, but when separated the monomeric subunits fail to display enzyme activity [1]. Each monomeric subunit of AP contains three divalent cations (two Zn2 + and one Mg2+) and a serine residue in the active site [2]. Previous studies on E. coli AP (ECAP) have shown that the two zinc ions are directly involved in catalysis (Fig. 2A) [2-4]. The two zinc ions are well positioned to activate the serine and water for nucleophilic attacks and they are involved in holding the phosphate moiety of substrate [2]. The Zn2+ ion at the first metal ion binding site (M1 site) is required for catalysis and plays an important role in binding both the substrate and the phosphate released upon hydrolysis (Fig. 2A). The second Zn2+ ion interacts with the hydroxyl group of the active site serine to stabilize the deprotonated form of the residue required for the nucleophilic attack on the phosphate [2]. Recent evidence suggests that the Mg2+ ion in the active site stabilizes the transferred phosphoryl group via a water molecule (Fig. 2A), and functions via a mechanism different from the two Zn2+ ions at the bimetallocentre [5].
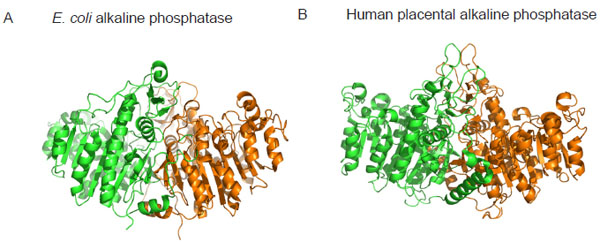
Crystal structure of alkaline phosphatase from E. coli (A) and human placenta (B) highlighting the structural similarity between bacterial and mammalian forms of the enzyme. Monomer subunits are coloured differently to highlight the dimeric nature of the enzymes. Coordinates of the structures were downloaded from http://www.rcsb.org/pdb and the images were produced using PyMOL [22]. The pdb accession numbers are ALK1 (E. coli alkaline phosphatase) and 1EW2 (human placental alkaline phosphatase).
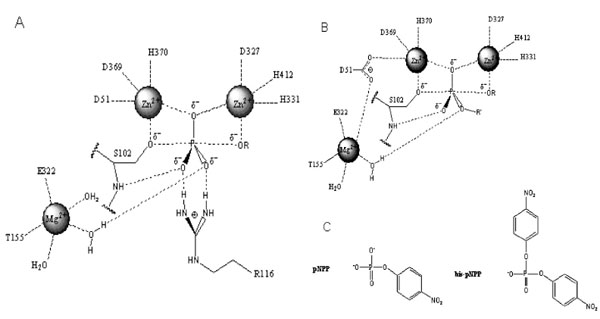
Structure of the active site and proposed reaction mechanism of alkaline phosphatase. (A). Schematic illustration of the active site and reaction mechanism of E. coli alkaline phosphatase. The bimetallocentre is occupied by the two Zn2+ ions, and Mg2+ ion at the third site is shown interacting with a non-bridging oxygen via a water molecule. (B). Proposed orientation of the R’ group of the diester substrate in the active site. The R’ group is oriented away from the Mg2+ site suggesting that hydrolysis of the diester substrate is less dependent on rate enhancement contribution from Mg2+. These figures are adapted from Zalatan et al., [5]. (C). Chemical structures of para-nitrophenyl phosphate (pNPP) and bis-para-nitrophenylphosphate (bis-pNPP) used as substrates for the monoesterase and diesterase activities of calf intestinal alkaline phosphatase.
APs have been classified into a superfamily of phospho-/sulfo-coordinating enzymes catalyzing the hydrolysis of phosphate monoesters, diesters, triesters, and sulfate esters [6]. The phosphodiesterase activity of AP has been demonstrated in the enzyme from rat osseous plate [7] and E. coli [6]. Mutational analysis suggests that common active site features contribute to hydrolysis of both phosphate monoesters and phosphate diesters. However mutation of the active site arginine to serine, R166S, decreases the phosphomonoesterase activity but not the diesterase activity, suggesting that interaction of this arginine with the nonbridging oxygen(s) of the phosphate monoester substrate is responsible for the preferential hydrolysis of phosphate monoesters [6].
Two-metal ion catalysis is a common feature of enzymes that catalyze phosphoryl transfer reactions including the hydrolysis of phosphate monoesters, diesters and triesters [8, 9]. The active sites of members of the alkaline phosphatase superfamily share several structural features underscored by the two-metal catalytic centre. Comparative analyses of the monoesterase and diesterase activities of APs are providing new insights into their catalytic mechanism [5, 7, 10].
The structural similarity shared by the active sites of related mono- and diesterases raises the possibility that any bimetallo active site might be able to catalyze these different classes of reactions. However, the partial negative charge on a phosphoryl oxygen atom of the phosphate monoester dianion is lost upon conversion to a diester monoanion, and the steric bulk of the added esterifying group of the diester could result in steric clashes within the active site [6].
Mammalian APs are found in several tissues including the intestine, kidney, liver and bone. Mammalian alkaline phosphatases perform a variety of functions including bone mineralization and fat absorption in the intestine, as well as vents in early embryogenesis [11]. These enzymes show structural similarity to ECAP (Fig. 1), and strong sequence conservation is seen in regions where the catalytic residues are located [12]. The APs from these sources act upon different phosphate monoesters and diesters. Hence, the mechanistic details of how mammalian APs deal with mono- and diester substrates is important to our understanding of the biological functions of the enzymes.
Divalent metal ions (such as Mg2+ and Zn2+) have both activating and inhibitory effects on mammalian alkaline phosphatase [1, 13, 14]. In an earlier report, we showed that optimal concentrations of both Mg2+ and Zn2+ were required for the tissue non-specific AP (TNAP) to catalyze the hydrolysis of the monoester substrate para-nitrophenyl phosphate [15]. Here, we investigate the effects of cofactor interactions on monoesterase and diesterase activities of calf intestinal AP (CIAP) with a view to gaining further insight into the interaction between the metal ions in AP catalysis.
MATERIALS AND METHODOLOGY
Reagents, Chemicals, and Enzyme
Alkaline phosphatase substrates, the sodium salts of para-nitrophenylphosphate (pNPP) and bis-para-nitrophenyl-phosphate (bis-pNPP), were obtained from Sigma-Aldrich, UK. Magnesium chloride (MgCl2) and zinc chloride (ZnCl2) were obtained from Fisher Scientific, UK. Homogenous CIAP was obtained from New England Biolabs, UK.
AP-Catalyzed Hydrolysis of pNPP and Bis-pNPP
Phosphomonoesterase and diesterase activities of CIAP were measured by the rate of hydrolysis of pNPP and bis-pNPP (Fig. 2C) respectively in the presence of the divalent metal ions Zn2+ and Mg2+ [7] at 37°C in 25 mM Tris-HCl (pH 8.5). Details of each experiment are described in the Figure Legends. Reaction mixtures containing CIAP and buffers in the presence of the appropriate metal ions were incubated at 37 °C for 10 minutes. Reactions were initiated by the addition of the appropriate concentration of the substrate. The absorbance was read at 410 nm against a blank of the buffered substrate. Spectrophotometric readings were taking in a Perkin Elmer Lambda 45 UV/VIS instrument and a Spectronic 21 spectrophotometer. All measurements of reaction rate were performed in triplicates.
RESULTS
Earlier studies on Escherichia coli AP (ECAP) showed that the enzyme is less efficient at hydrolyzing phosphodiester substrates than phosphomonoesters [6]. Here, we compared the rates of hydrolysis of pNPP and bis-pNPP by CIAP using a simple time-course analysis. Both reactions were studied under identical conditions of 2.5 mM substrate concentration. This concentration is sufficient to saturate 10 µM CIAP used in these reactions [14, 15]. Hydrolysis of pNPP in the monoesterase reaction was monitored spectrophotometrically over 15 minutes, while the slower phosphodiester hydrolysis of bis-pNPP was followed for 180 minutes. The rates of both reactions were linear over the time ranges studied. The monoesterase activity (0.0383 min-1) was 30 times faster than the diesterase activity (0.0013 min-1) of CIAP under these conditions (Fig. 3A). These results show that the CIAP active site is better adapted to deal with monoesters than phosphodiester substrates, and are in agreement with earlier studies on related members of the alkaline phosphatase superfamily [6, 16].
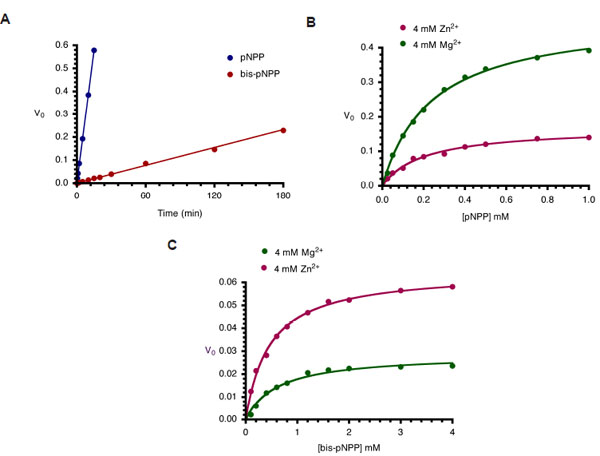
Kinetics of monoesterase and diesterase activities of CIAP. (A). Time course of hydrolysis of pNPP and bis-pNPP. Reaction mixtures (200 µl) containing 25 mM Tris-HCl (pH 8.5), 2.5 mM p-nitrophenyl phosphate (or bis-p-nitrophenyl phosphate), 100 µM ZnCl2, 1 mM MgCl2, and 10 µM CIAP were incubated at 37 °C for the indicated time. Reactions were initiated by the addition of the substrate to the reaction mixture and stopped by the addition of 1.0 ml 0.5 M NaOH. Activities are expressed as change in A410 per minute for phosphomonoesterase activity and change in A410 per hour for the phosphodiesterase activity. (B) Effects of Zn2+ and Mg2+ on kinetics of CIAP-catalyzed hydrolysis of pNPP. The initial rates of hydrolysis of increasing concentrations of pNPP by 10 µM CIAP at 37 °C in the presence of 4 mM Zn2+ or 4 mM Mg2+ were determined. The data were fitted to the Michaelis-Menten curve using GraphPad Prism curvefitting software. The concentrations of pNPP used were 0.025, 0.05, 0.1, 0.15, 0.2, 0.3, 0.4, 0.5, 0.75, and 1.0 mM. (C) Effects of Zn2+ and Mg2+ on kinetics of CIAP-catalyzed hydrolysis of bis-pNPP. Reactions and data analysis were carried out as described in (B). The concentrations of bis-pNPP used were 0.1, 0.2, 0.4, 0.6, 0.8, 1.2, 1.6, 2.0, 3.0, and 4.0 mM.
In order to characterize the metal ion preference of CIAP for the two activities, we compared the effects of Zn2+ and Mg2+ on the hydrolysis of pNPP and bis-pNPP. CIAP was incubated with 4 mM Mg2+ or Zn2+ at 37 °C for 10 minutes to ensure binding to the metal ion sites of the enzyme [15]. The cofactor-saturated CIAP were added to reaction mixtures containing increasing concentrations of pNPP and bis-pNPP. Initial rates were determined over linear ranges similar to those shown in Fig. (3A), and the values were plotted as Michaelis-Menten curves in Figs. (3B and 3C). The results show that the two metal ions manifest opposite effects on monoesterase and diesterase activities of CIAP. Kinetic constants calculated from the pNPP reaction show that Mg2+ (Vmax 0.493 min-1, Km 0.239 mM, K0 2.062) was a better activator than Zn2+ (Vmax 0.169 min-1, Km 0.201 mM, K0 0.841). In contrast, Zn2+ (Vmax 0.065 min-1, Km 0.459 mM, K0 0.141) was preferred to Mg2+ (Vmax 0.029 min-1, Km 0.607 mM, K0 0.047) in the hydrolysis of bis-pNPP by CIAP.
Next, we examined the effects of increasing cofactor concentrations (Zn2+ and Mg2+) on the monoesterase and diesterase activities of CIAP. Aliquots of CIAP were incubated with 0 to 10 mM Mg2+ and/or Zn2+ at 37 °C for 10 minutes as described earlier. The effect of Zn2+ was biphasic as the activation effect peaked, and increasing concentration resulted in a steady decrease in activity. In contrast, Mg2+ at equivalent concentration led to continued activity of the monoesterase reaction. The effects of Mg2+ and Zn2+ on the diesterase activity of CIAP (Fig. 4B) were different from the pattern seen in the monoesterase reaction (Fig. 4B). Zn2+ was a better activator of bis-pNPP hydrolysis, and only marginal stimulation of activity resulted from pre-incubation of the enzyme with Mg2+. Higher concentration (10 mM) of Zn2+ did not inhibit bis-pNPP (Fig. 4B) hydrolysis in contrast to the inhibition caused by Zn2+ concentration higher than 2 mM in the pNPP reaction (Fig. 4A).
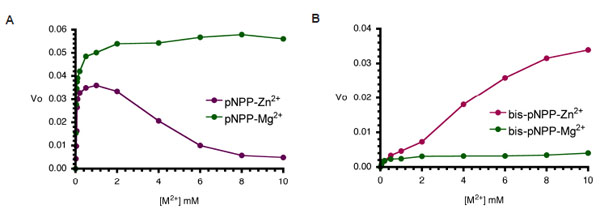
Metal ion dependence of CIAP-catalysed hydrolysis of pNPP and bis-pNPP. Reaction mixtures (200 µl) containing 25 mM Tris- HCl (pH 8.5), 2.5 mM p-nitrophenyl phosphate (A), or bis-p-nitrophenyl phosphate (B), the indicated concentration of the metal ion (ZnCl2 or MgCl2), and 10 µM CIAP were incubated at 37 °C for 10 minutes. Reactions were initiated by the addition of the substrate to the reaction mixture and stopped by the addition of 1.0 ml 0.5 M NaOH. Activities are expressed as change in A410 per minute for phosphomonoesterase activity and change in A410 per hour for the phosphodiesterase activity.
AP-catalysed hydrolysis of pNPP is activated by both Mg2+ and Zn2+ ions in a way characteristic of synergistic interactions [15]. Since, optimal monoesterase activity of AP requires the interplay of both Zn2+ and Mg2+ ions, we compared the monoesterase and diesterase activities of CIAP in the presence of the two divalent metal ions (Fig. 5A and 5B). In agreement with earlier findings [14, 15], both metal ions are required for efficient monoester (pNPP) hydrolysis (Fig. 5A). However, the Zn2+-ion activated hydrolysis of bis-pNPP by CIAP was inhibited when increasing concentrations of Mg2+ ion were added to the reaction (Fig. 5B). Zn2+ ion is the preferred cofactor for the phosphodiesterase activity of CIAP, which is less dependent on Mg2+. Indeed, addition of Mg2+ specifically inhibited the hydrolysis of bis-pNPP.
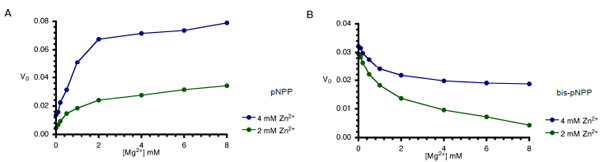
Effects of Mg2+ on Zn2+-activated hydrolysis of pNPP and bis-pNPP by CIAP. Reaction mixtures (200 µl) containing 25 mM Tris- HCl (pH 8.5), 2.5 mM p-nitrophenyl phosphate (A), or bis-p-nitrophenyl phosphate (B), 2 mM or 4 mM ZnCl2 plus the indicated concentration of MgCl2 (0-10 mM), and 10 µM CIAP were incubated at 37 °C for 10 minutes. Reactions were initiated by the addition of the substrate to the reaction mixture and stopped by the addition of 1.0 ml 0.5 M NaOH. Activities are expressed as change in A410 per minute for phosphomonoesterase activity and change in A410 per hour for the phosphodiesterase activity.
DISCUSSION
Our finding in this study that the phosphomonoesterase activity of CIAP is more efficient than its phosphodiesterase activity reflects similar reports on ECAP [5]. Other mechanistic differences have been reported between the monoesterase and diesterase functionalities of ECAP. O’Brien et al., [17] reported that the R166S mutation in ECAP specifically inhibited monoesterase activity without affecting the hydrolysis of diester substrates, suggesting that the residue is important for binding and positioning of monoester substrates. The lack of effect of the R166S mutation on diester hydrolysis shows that this interaction does not contribute to the reaction of phosphate diesters, presumably because the introduction of steric bulk caused by esterification of a non-bridging oxygen of the transferred phosphoryl group prevents this interaction with the phosphate diester substrate [6].
Zalatan et al., [5] showed that removal of the Mg2+ site in ECAP had no significant effect on the diesterase reaction, while causing a significant decrease in monoesterase activity. Structural analysis and comparative studies with sulphate monoester substrates show that the R’ group of diester substrates is oriented away from the Mg2+ site in AP (Fig. 2B). Hence, the active site Mg2+ has little or no contribution to rate acceleration of diester hydrolysis, in contrast to the monoesterase reaction that depends on Mg2+ in transition state stabilisation (Fig. 2A). It follows that catalysis of the diesterase reaction relies more on interactions with the bimetallocentre occupied by the two Zn2+ ions. This unique property of diesterase catalysis by AP might explain the inhibitory effect of Mg2+ if it displaces Zn2+ from the bimetallocentre [15]. In a comparative analysis of the hydrolysis of several diester substrates by ECAP-R166S, Nikolic-Hughes et al., [18] showed that rate enhancements correlated with the amount of negative charge localised between the two Zn2+ ions in the active site. Hence, the electrostatic features of the active site can be tuned to favour monoesterase or diesterase reactions when the coordinating properties of the bimetallocentres are altered. Such electrostatic contributions to the catalysis of diester hydrolysis by CIAP would be perturbed by the introduction of Mg2+ into one or both of the Zn2+ catalytic sites.
The synergistic effect of Mg2+ and Zn2+ in the activation of phosphomonoesterase activity of AP observed in this study confirmed our earlier report of the synergistic effects of Zn2+ and Mg2+ ions in the activation of TNAP [14, 15]. These and related findings [19, 20] suggest that the synergistic effect of the two cofactors is a recurrent theme of the two-metal ion mechanism of the alkaline phosphatases. However, the effect of interaction of Mg2+ and Zn2+ in the hydrolysis of bis-pNPP by CIAP as observed here did not show any synergistic effect though each metal ion in the absence of the other activates the enzyme. This would not be surprising if Mg2+ makes little contribution to rate enhancement of diester hydrolysis [5], in contrast to its significant impact in hydrolysis of phosphomonoester by alkaline phosphatase [15]. Activation of phosphodiesterase activity of CIAP by Mg2+ was limited, Zn2+ being the preferred metal ion for the hydrolysis of bis-pNPP. Interestingly, Mg2+ seems to displace Zn2+ from the enzyme and significantly inhibits the diesterase reaction. Keppetipola and Shuman [21] described mutants of a polynucleotide kinase/phosphatase from Clostridium temocellum with different cofactor requirements for its monoesterase and diesterase activities.
The alkaline phosphatase superfamily of hydrolase enzymes exhibits a high degree of substrate flexibility. This broad specificity means that the enzymes could be engineered to act with different affinities on a variety of phosphate esters. This can be achieved via random or directed mutagenesis of active site residues. The findings from this study offer the prospect of altering the substrate specificities of alkaline phosphatases and related bimetalloenzymes through changes in reaction cofactors in addition to efforts resulting from active site engineering.
CONFLICT OF INTEREST
None declared.
ACKNOWLEDGEMENTS
We are grateful to Dr. Adrian Lapthorn (University of Glasgow) for generating Chemdraw formats of chemical structures; and Drs Sean Colloms and Louise Horsfall (University of Glasgow) for their helpful comments on the manuscript.
REFERENCES
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]

