All published articles of this journal are available on ScienceDirect.

Influence of Aromatic and Aliphatic Moieties on Thrombin Inhibitors Potency
Authors Info & Affiliations
Abstract
Thrombin is a plasma serine protease that plays a key role in coagulation and hemostasis but also in thromboembolic diseases. Direct thrombin inhibitors could be beneficial for future anticoagulant therapy in the prophylaxis of venous and arterial thrombosis as well as myocardial infarction. To design the efficient thrombin inhibitors we have synthesized and studied peptide-based inhibitors resistant to enzymatic degradation. Compounds with general formula X-DArg-D-Phe-OMe, where X = residue of 3-[6-ethyl-7-hydroxy-3-(4-methyl-thiazol-2-yl)-4-oxo-4H-chromen-2-yl]-propionic acid (chromone) and lauric acid were synthesized by classic methods of peptides synthesis in solution. The comparative inhibitory analysis of prepared compounds in relation to thrombin was conducted. The analysis of the inhibition effect of the peptide with retro-D-sequence modified by residues of natural organic compounds (chromone or fatty acid moiety) has demonstrated that modification with the fatty acid residue appeared to be the most successful one. Introduction of lauric acid residue (Ki = 1,76 μM) maximally increased the inhibition effect. These findings establish an important role of fatty moiety in structure of inhibitors in preferential binding and inhibition of thrombin active side.
INTRODUCTION
Thromboses and thromboembolic diseases are the most widespread in developed countries. In present medical practice the natural thrombin inhibitor – heparin and its low-molecular fragments are widely used, which act by formation of the triple complex with thrombin and antithrombin III– as anticoagulants of direct action (Mousa SA. In Methods Mol. Med. 2003) [1] (Smith Thromb. Res. 1981) [2]. Warfarin is a coumarin derivate which is used as the anticoagulant of indirect action, an antagonist of synthesis of vitamin K dependent enzymes, which include the precursor of thrombin – prothrombin (O’Reilly In Hemost. Thromb. 1987) [3]. Several pharmaceutical companies have therefore very intensely pursued the search for low molecular weight active site thrombin inhibitors over the past years. However, development of direct thrombin inhibitors has brought researchers more “headache” than success. The most recent setback came when AstraZeneca withdrew Ximelagatran, the first orally bioavailable direct thrombin inhibitor that had received regulatory approval, after reports of a significant risk of potentially lethal hepatic toxicity in a fraction of patients (Testa Int. J. Cardiol. 2007) [4].
Thrombin (EC 3.4.21.5), the key enzyme of the hemostasis system, is the therapeutic target to create anticoagulants of direct action – effective pro-drugs for treatment of thromboembolic diseases (SandersonCurrent Medical Chemistry 1998) [5] (Bauer Hematology2006) [6]. Structures of the
active site of thrombin as well as numerous functions are currently well studied (Fenton Blood Coagu. Fibrino. 1991) [7] (Bode Journal of Thrombosis and Haemostasis 2005) [8] (Stubbs Thromb. Res. 1993) [9] (Huntington J.Thromb. Haemos. 2005) [10]. X-ray structural analysis data of complexes of thrombin with inhibitors enabled to design new inhibitors of the enzyme with high efficiency (Bode A Protein Science 1992) [11].
The special attention is devoted to selectivity of inhibitors, which can be effectively provided by substances of the peptide nature. Unfortunately, recently this class of compounds has not been causing an interest because peptides are characterized by extraordinarily low bioaccumulation due to non-specific degradation by proteases that reduce an inhibitor delivery probability to chosen target enzyme. To prolong the lifetime of low-molecular peptide inhibitors of thrombin, that is to make them invulnerable to non-specific hydrolysis by proteases, we have applied to design them topochemical approach introduced by Shemyakin, Ovchinnikov and Ivanov (Shemyakin Angew. Chem. 1969) [12]. The key point of this approach consists in synthesis of retro-D-analogues of natural biologically active peptides. Peptides with the retro-D-sequence are resistant to enzymatic degradation of the reversed amide bond and preserve ability to binding with the proper active sites of enzyme.
However, this approach has been productive only for cyclopeptide compounds, but linear retro-D-analogues preserving a resistance to enzymatic hydrolysis had low biologic activity. We decided to modify this approach by insertion of natural compound residue into the peptide motif in order to intensify the biologic effect of compounds.
Many thrombin inhibitors share the D-Phe-Pro-Arg motif mimicking the binding region of the natural substrate fibrinogen (Bajusz J. Med. Chem. 1990) [13] (Brady Bioorg. & Med. Chem. 1995) [14] (Bajusz Bioorg. Med. Chem. 1995) [15]. A number of known inhibitors such as inogatran and melagatran (active form of ximelagatran) have been developed based on this motif (Preville Bioorg. Med. Chem. Lett.1997) [16] (Gustafsson Thromb. Res. 2001) [17]. Peptidomimetic thrombin inhibitors with different modifications by isosteric amino acids of this thrombin inhibiting tripeptide chain were also synthesised (Nöteberg. Med. Chem. 2000) [18] (Throstensson J. Med. Chem. 2003) [19].
Notwithstanding, we have demonstrated that dipeptide Tos-Phe-Arg-OCH3 is both the substrate and inhibitor of thrombin and has an (IC50 = 20 μM) appropriate characteristics to be transformed into the retro-D-sequence with efficient binding in the active site of thrombin (Poyarkova Ukrainskii Biokhimicheskii Zhurnal 1986) [20]. Retro-D- sequence of this dipeptide D-Arg-D-Phe was successfully used as ligand of the affinity sorbent for highly purified thrombin preparation with the activity of 6000 NIH Е/mg (Haga SUPatent 1527261, 1989) [21]. Therefore we used D-Arg-D-Phe motif to create a new thrombin inhibitors resistant to enzyme degradation and carried forward biological active blocks which are useful for others steps of cascade coagulation.
There are no data about synthesis and inhibitory effect of retro-D-peptides, modified by natural aromatic and fatty acid moieties. In this study we synthesized, investigated antithrombotic activity and compared inhibitory activity of new thrombin inhibitors high-resistant to enzymatic digestion. This study provides new insights into the role and effect of fatty acid tail on the interaction behaviors with the secondary binding site of thrombin and is important for future development of new anticoagulants, non toxic and resistant to enzymatic degradation.
MATERIALS AND METHODS
Amino acids, fatty acids and solvents were pursued from "Merck" (Germany), condensing agents - from “Sigma” (Germany). The reaction control and purity of the products were monitored by TLC on plates with Silica gel 60 F254 (“Merck” Germany) (n-butanol/acetic acid/water, 4:1:1, v/v (A); benzene/ethyl acetate, 5:4, v/v (B); chloroform/methanol, 8:2, v/v (C). After chromatography the plates were developed with ninhydrin, iodine, Sakaguchi reagent or ammonium molybdate. rpHPLC separation was conducted on Waters chromatography system equipped with two 510 Waters High pressure pumps, 996 Waters PDA detector with base wavelength 214 nm.
The molecular masses were analyzed by LDI mass spectrometry on Bruker AUTOFLEX II LRF20 spectrometer.
1H NMR spectra were registered on a Varian VXC-400 (Germany) (400 MHz) in DMSO- d6 (99.9%) relative to residual solvent protons.
Methods of Synthesis
Methyl ester of carbobenzoxy-D-arginyl-D-phenylala-nine (1) was synthesized as described in the work (Haga SUPatent 1527261, 1989) [21].
A mixture of Z-D-Arg (4,51g, 14,6 mmol) and D-Phe-OMe (3,2g, 14,8 mmol) was dissolved in 30 ml of absolute pyridine and was stirred for 30 min, then was cooled to 0°C. DCC (3,06 g, 14,8 mmol) was added to the cold solution. The reaction mixture was stirred for 6 hours at 0°C and kept for 24 hours at room temperature. The precipitate obtained was filtered, and the solvent was evaporated under vacuum at 40°C. The residue was dissolved in butanol-2 and washed with the saturated solution of NaCl and 1N HCl. Butanol-2 was evaporated, and the residue was crystallized from the mixture of methanol – ethyl acetate. The yield 1 was 5, 84 g (92%); mp 80-98°C; Rf = 0,65 in the system (A). Data of the element analysis: Found %: С = 57, 21; Н = 6,17; N = 13,64. С24Н31N5О5.HCl.H2O; calcd %: С = 56,97; Н = 6,18; N = 13,84.
Methyl ester of N( - lauroyl-D-arginyl-D-phenylalanine (3) was similarly synthesized from 0,5 g (1,27 mmol) of monohydrochloride of lauroyl-D-arginine [31], 0,27g (1,27 mmol) of D- phenylalanine methyl ester and DCC 0,27 g (1,30 mmol). The product obtained was crystallized from ethyl acetate, 0,5g (70%); mp 92-93˚С; Rf = 0,43 in the system (B). Data of the element analysis: Found %: С =61.01; Н = 9.01; N =12,28. С28Н47N5О4 х HCl ; calcd %: С =60,69; Н = 8,73; N =12,64. HPMS m/z 518,083 (М + Н)+ calcd for C28H47N5O4 517, 70
N-oxysuccinimide ester 3-[6-ethyl-7-hydroxy-3-(4-methyl-thiazol-2-yl)-4-oxo-4H-chromen-2-yl]-- propanoic acid 0,4 g (1 mmol) of 3-[6-ethyl-7-hydroxy-3-(4-methylthiazol-2-yl)-4-oxo-4H-chromen-2-yl]-propanoic acid synthesized as described in the work (Poyarkov Russian J. Bioorg. Chem. 2005) [22] were dissolved in 20 ml of distilled DMF, and at cooling to 4°C N-hydroxysuccinimide (0,12 g, 1,01 mmol) and DCC (0,21g, 1,01 mmol) were added. The mixture was stirred for 12 hours at 4°C. After reaction completion (control – TLC) the precipitate DCU was filtered, DMF was evaporated in vacuum at 40°C. The residue was crystallized from iso-propanol; 0,4g (80%); mp 110°; Rf=0.6, (B) benzene ethyl-acetate (5:4).
Methyl ester of D-arginyl-D-phenylalanine (1a) 1 ml of 5,6 N HCl and 1 g of catalyst Pd/C was added to solution of 5,84 g (1) in 30 ml of methanol, and hydrogen was passed through the suspension for 10 hours at the ambient temperature and stirring. The catalyst was filtered, a solvent was evaporated, and the precipitate was crystallized from absolute ether. Yield 4,37 g (93%); mp 148-152° C; Rf = 0,15 (A) (4:1:1).
Methyl ester 3-[6-ethyl-7-hydroxy-3-(4-methyl-thiazol-2-yl)-4-oxo-4H-chromen-2-yl]-propionyl-propionyl-D- arginyl-D-phenylalanine). (2) To solution of 0,3 g (0,735 mmol) of D-arginyl-D-phenylalanine methyl ester hydrochloride in 20 ml of DMF 0,074 ml of N-methylmorpholine were added and the solution was stirred for 20 min. at room temperature, the precipitate of N-methylmorpholine hydrochloride was filtered. N-oxysuccinimide ester of 3-3-[6-ethyl-7-hydroxy-3-(4-methyl-thiazol-2-yl)-4-oxo-4H-chromen-2-yl]-propionic acid (0,3350 g, 0,735 mmol) was added to the filtrate. The reaction mixture was stirred at ambient temperature for 48 hours, then the solvent was evaporated and the residue was crystallized from ether. The product was purified by rpHPLC on the preparatory “Cromasil C18” column (10x250 mm) using aqueous buffers as the mobile phase – phase A (10% CH3CN and 0,1% of TFA) and phase B (with 80% CH3CN), respectively. The separation was conducted in conditions of linear gradient elution. The retention time of the target fraction was 27,7 min. Solvent of the target faction was evaporated, and the residue was crystallized from ether. Yield 0,2 g (36%); mp 160ºC; Rf = 0,33 system (A) (4:1:1). 1H NMR (400 MHz, DMSO-d6) : 8,34-8,33 (1 H, d, J 4); 8,08-8,06 (1 H, d, J 8); 7,8 (1 H, s,); 7,77 (1H, s,); 7,3 (5 H , m.); 7,19 (1 H, s,); 6,95 (1 H, s,); 4,49 (1 H m, J 6); 4,36 (1 H, m, J 8,); 3,61 (1 H, s,); 3,60 (5 H, m.); 3,55 (3 H, s.); 2,95 - 2,97 (2H, m.); 2,62 - 2,7 (2 H, m); 2,45 (3 H , s); 1,3 - 1,7 (4 H, m); 1,24 (3 H, t. J 8,9).
Thrombin Assays
Inhibition of the Thrombin Coagulating Activity (IC50 Determination)
Preparation of fibrinogen solution as well as buffer solutions for determination of coagulant activity of thrombin were carried out in accordance with the method (Baughman In Methods Enzymol. 1970)[23]. The starting solution of human thrombin isolated using the Fenton method (activity 3000 NIH E/mg) was prepared so that the time of coagulating of 0,1 % solution of fibrinogen (Serva, 90% of coagulated protein) made up 15-20 sec. under standard conditions (Fenton J. Biol. Chem. 1977) [24]. The study of the investigated compounds on coagulant activity of thrombin was conducted at the following conditions. 0,04 M Tris-HCl buffer (pH 7,3) containing an inhibitor, 0,15 M NaCl and 0,66% PEG 6000 (Serva) was added to 0,2% solution of fibrinogen (0,5ml) in the same buffer. The reaction mixture was thermo stated for 5 min. at 29°C, then treated with the starting thrombin solution (20 μl), and the time needed for clot formation was measured. The time of fibrinogen coagulating was determined three times for 5-6 various concentrations of peptide. The value of ІC50 was calculated from the curve (τ-τ0)/ τ0 depend of [I], where τ0 is the time of fibrinogen coagulating at absence of inhibitor; τ is time of fibrinogen coagulating at presence of the explored compound; [I] – concentration of inhibitor.
Chromogenic Assays
Inhibition of the Thrombin Amidolytic Activity
The inhibition constant (Ki) for dissociation of the enzyme-inhibitor complex was determined by a study the impact of the inhibitor concentration on initial rate of p-nitroaniline elimination from the chromogenic substrate of Tos-Gly-Pro-Arg-pNA (Chromozym TH). Change of rate of the enzymatic reaction was determined based on growth of p-nitroaniline absorption at the wavelength of 405 nm using a microtiter plate reader (Vmax Microplate Reader. Molecular Devices. USA) at the ambient temperature in the medum of 0,05M Tris/HC1 buffer of pH 8,0 containing 0,15 M NaCl and 0,66% PEG 6000. Compound 2 was studied in the same buffer containing 3% DMSO in final concentration.
Assays were performed according to the following general procedure. The solution of human thrombin (1.67 nM) was added to the solution containing an inhibitor (varying concentrations, from 2 to 200 μМ), incubated for 5 min. and the reaction was initiated by adding the substrate (250 μМ), increase of absorption was registered every minute during 3 min.
The data obtained were calculated in the Dixon system of co-ordinates (1/V( [I]) using EXCEL software. IC50 was defined from the linear dependence equation у=ах+b, as x =-(b/a), where y=1/V, and x= [I] inhibitor concentration. The Ki value was calculated from the IC50 value using the equation, Ki = IC50/(1+[S]/Km) assuming a competitive enzyme inhibition.
The Km (26,3±0,18) μМ value for (Chromozym TH) was defined at the same conditions at various concentrations of the substrate and calculated in double reverse co-ordinates of Lineweaver – Berk. The constants were determined based on three independent experiments. The experiment standard deviation did not exceed 10%.
RESULTS
To construct structures of desired compounds, we selected N-terminal substituents based on the following criteria:
Compounds containing the chromone nucleus are universal pharmacophores (depending on substituents). (Mogilevich. In Chem. Bioregul. Proc. 1992) [25]. Moreover, dicoumarin (warfarin) is known to be the indirect anticoagulant, which suppresses synthesis of vitamin K dependent proteins, including the precursor of thrombin – prothrombin (Smith Thromb. Res. 1981) [2]. Besides, recently it was discovered that flavonoids inhibit the amidolytic activity of human thrombin (Mozzicafreddo Biochimie 2006) [26]. Therefore it is possible to assume that inhibitor degradation products, like coumarin analog substituent by itself, will make an impact on earlier stages of the blood coagulation cascade.
Fatty acids perform extraordinarily important functions in an organism, and their derivatives are supposed to possess the ability to penetrate through cells membranes that increases a probability of inhibitor transportation to the chosen target (Leung J. Med. Chem. 2000) [27].
N-α-Lauroyl-L-arginine ethyl ester monochloride (LAE) is being developed as a novel preservative in selected foods and is being produced by Laboratorios Miret S.A. (LAMIRSA). It was shown that LAE possessed a very low toxicity and high antimicrobial activity (Ruckman Food Chem. Toxicol. 2004) [28], (Rodríguez J. Appl. Microbiol. 2004) [29].
The retro-D-sequence D-Arg-D-Phe was chosen as a peptide motif because of affinity toward thrombin (Haga SU Patent 1527261, 1989) [21]. Involving into this sequence aromatic or aliphatic (in general hydrophobic) moieties increases the affinity to enzyme by supplementary interactions with the hydrophobic region of thrombin, immediately adjacent to the active site. For this purpose we have developed appropriate synthetic methods for introduction of the aromatic or aliphatic moieties to D-peptides sequence.
Chemistry
Modification of D-arginine on the α-amino group with biologically active compounds was carried out using N-oxysuccinimide esters of chromone as described in (Poyarkov Russian J. Bioorg. Chem. 2005) [22]. N-Oxysuccinimide esters, along with their high reactivity, are resistant to hydrolysis, which makes it possible to conduct synthesis in aqueous dioxane. Use of N-oxysuccinimide esters of acid derivatives of chromone and fatty acids enabled to obtain arginine derivatives in high yield and in some cases without extraction of intermediate N-oxysuccinimide esters of original acids. The derivatives with C-terminal free arginine were easily purified by crystallization from water ethanol.
Carboxyl function of phenylalanine was protected by esterification with methyl alcohol. Condensation of protected amino acids was mainly conducted by the carbodiimide method in pyridine using protonated protection of the guanidine function of arginine. To obtain the chromone derivative we used condensation of the corresponding N-oxysuccinimide ester with hydrochloride of D-arginyl-D-phenylalanine methyl ester in DMF, as indicated in Fig. (1). Purification of the compound Chrom-D-Arg-D-Phe-OMe was conducted by the rpHPLC method. Purity of the compound obtained is proven by the NMR.
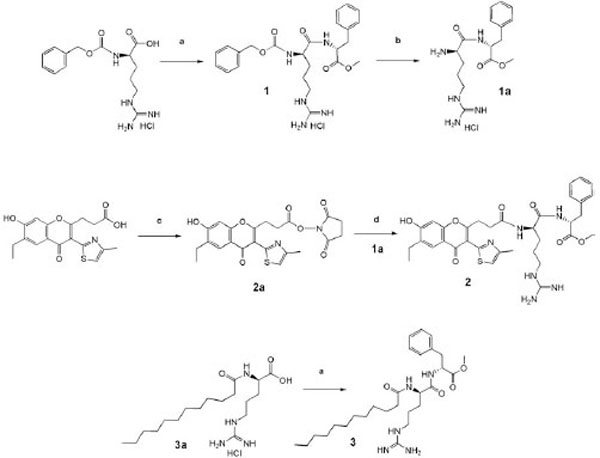
Synthesis of the products. Reagents and conditions: a) D-Phe-OMe, DCCD /Pyridine; (b) H2/Pd, methanol; (c) HONSu, DCCD /DMFA; (d) 1a, N-methylmorpholine / DMFA.
Biological Activity
Biological activity of the synthesized compounds was studied by their ability to prolong the time of fibrinogen coagulation by thrombin and by inhibition of the thrombin's cleavage reaction of the chromogenic substrate of Tos-Gly-Pro-Arg-pNA (Chromozym TH). (Table 1)
Inhibitory Activity of the Retro -D-Sequence of DArg-D-Phe Peptides - Modified on the α-Amino Group of Arginine
| № | Compounds | IC50 µM | Ki µM |
|---|---|---|---|
| 1 | Cbz-D-Arg-D-Phe-OMe | 110 | 42± 5 |
| 2 | Chrom-D-Arg-D-Phe-OMe* | 56 | 14,35 ± 0,92 |
| 3 | Laur-D-Arg-D-Phe-OMe | -//- | 1,76 ± 0,09 |
* Inhibitory effect was studied in the same buffer containing 3% DMSO in final concentration.
Measurements were conducted according to standard procedure which is carefully described in materials and methods.
To estimate the anticoagulation action of synthesized compounds we explored a capability of these compounds to increase the time of formation of the fibrin clot at thrombin impact (Baughman In Methods Enzymol. 1970)[23]. As it follows from the IC50 values given in Table 1, compound 2 possesses the maximal inhibition effect. The compound Cbz-D-Arg-D-Phe-OMe (1), which has been already used for synthesis of the affined absorbent and as the started compound for synthesis of the chromone derivative, also possesses anti-thrombin activity (IC50=110μM) which is twice lower than the corresponding activity of compound 2 that it indicates insufficient affinity of this compound to the active site of thrombin. Unfortunately, we could not determine IC50 for compound 3, due to its capability to precipitate the fibrinogen from its solution, this property needs additional study. Nevertheless it will be useful to decrease the fibrinogen concentration and to inhibit blood platelets aggregation. This process will be beneficial to prevent the risk of cardio-vascular diseases.
Thrombin inhibition by the compounds was studied by their ability to reduce a rate of hydrolysis of Chromozym TH in the conditions of steady-state kinetics at [S] > 10 Km. As it follows from the data presented in Table 1, the maximal inhibition effect on amidolythic activity of thrombin is observed for compound Laur-D-Arg-D-Phe-OMe 3 modified with the residue of lauric acid. The compound Chrom-D-Arg-D-Phe-OMe 2 containing in its structure the chromone residue demonstrated a considerably worse inhibition effect. Definition of its activity was carried out in the same Tris HCl buffer containing 3% of DMSO due to its low solubility in water. The benzyloxycarbonyl derivative of dipeptide 1 is characterized by the minimum inhibition effect.
DISCUSSION
The surprisingly low value of Ki of the compound 3 modified with the lauric acid residue motivated us to conduct additional researches to get an understanding of the interaction of such compound at the binding site of thrombin. Nα-lauroyl-L-arginine ethyl ester (LAE) (partial L-analog of compound 3 is well known surfactant (Infante Fette Seifen Anstrichmittel. 1985) [30]). Nevertheless we studied inhibition effect of LAE earlier and showed that this compound selectively inhibits thrombin activity (Ki=2,0 µM) almost in 20 times more strongly than trypsin (Кі=19,0 µМ). At the same time LAE maintain capability to be hydrolyzed by thrombin at pH 8,5 (kcat = 3,6 с-1) and trypsin (kcat = 56 с-1) (Poyarkov Ukrainskii Biokhimicheskii Zhurnal. 2007) [31].
If the LAE interaction mechanism in the active center of thrombin is well-defined, the interactions of substituents of retro-D-chain are not absolutely clear. That is why the supplemental experiments have been made. Studying of inhibition effect of Laur-DArg-DPhe-OMe on thrombin at different substrate concentrations has shown the competitive character of inhibition Fig. (2) according to (Cornish-Bowden In Principles of Enzyme Kinetics 1976) [32].
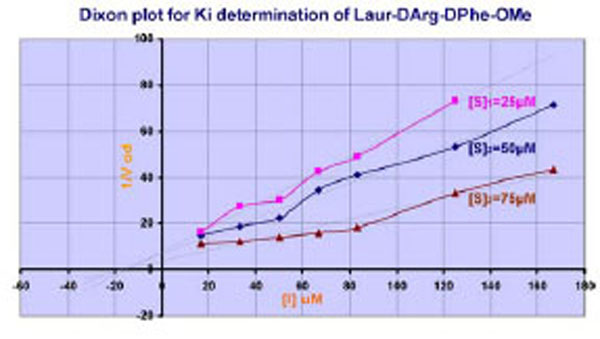
Plot for Ki estimation of Laur-DArg-D-Phe-OMe. [S]1,[S]2, and [S]3 are appropriate Chromozyme TH concentrations.
In this case, it is not quite appropriate to use the scheme by Schechter-Berger (Schechter Berger Biochem Biophys Res. Commun. 1967)[33] and it is more reasonable to consider co-reaction with the active site of thrombin taking into account the structure of the secondary binding center of thrombin (Bode EMBO J. 1989) [34]. According to the data of X-ray analysis, the structure of the secondary binding site of thrombin is organized as so-called «hydrophobic cage» formed by the following residues of amino acid: Ile174, Trp215, Leu99, His57, Tyr60A and Trp60D. The special folding of thrombin ( chain in the form of a stretched loop with Tyr60A and Trp60D located together with the Trp148 forms a narrow and deep cleft of the active site. The indole rings of Trp60 D and Trp148 cover the cleft, thus hampering access to the active site of thrombin.
Taking into account that a length of synthesized peptides is limited by two amino acid residues and they have not rigid conformation, it seems logical to assume that the fatty tail of lauric acid appears engaged in cooperation with D-phenylalanine through the hydrophobic effect and located in the «hydrophobic cage» of the secondary binding site of thrombin. The authors of the article (Lau Bioorg. Med. Chem. 1995) [35] make a schematic diagram of active site showing key binding features of thrombin. In comparison the binding of our compounds with the known anticoagulant agratroban, we used this scheme to illustrate the occupation of the Laur-D-Arg-D-Phe-OMe in the active site of thrombin as it is shown in Fig. (3).
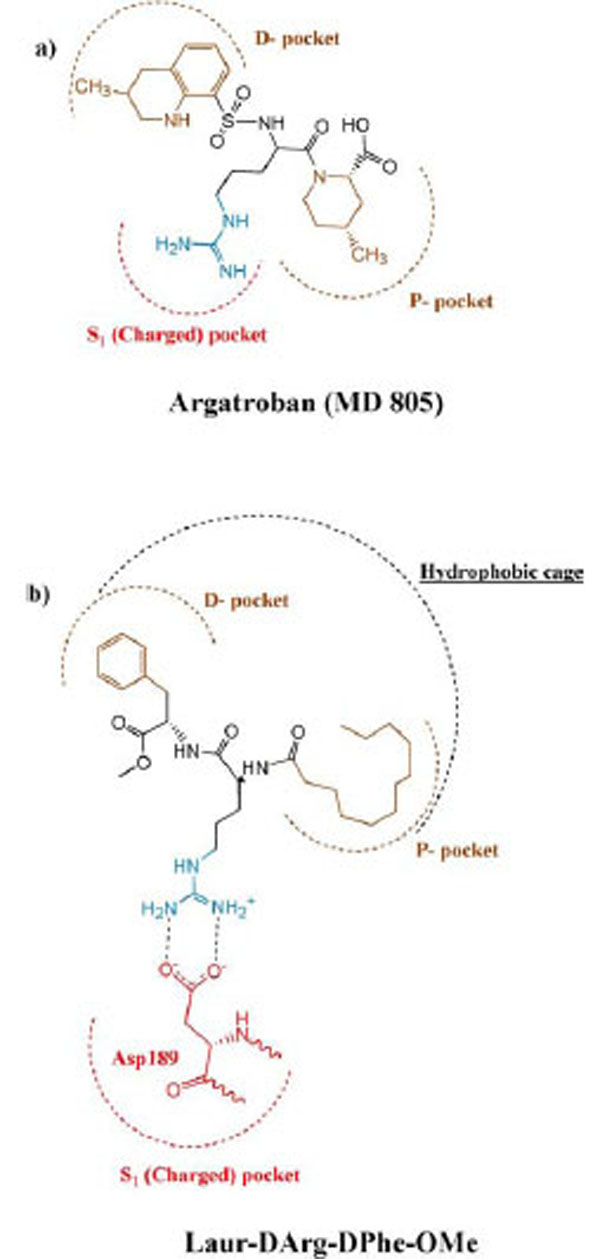
Schematic representation of the binding mode of argatroban (MD 805) (a), and Laur-DArg-DPhe-OMe (b) to the active site of thrombin according to (Lau, Bioorg. Med. Chem. 1995)[35].
Argatroban has hydrophobic moieties that bind in the hydrophobic “P-pocket” and the “D-pocket, and a guanidinium moiety that binds in the specificity pocket and forms a salt bridge with the carboxyl group of Asp 189. “P-pocket” and “D-pocket as used by Banner and Hadvary (Banner J Biol Chem 1991) [36]. The Tyr60A-Pro60B-Pro60C-Trp-60D loop creates with Trp215. Leu99, and Ile174 a double hydrophobis pocket which they have divided into the P-pocket (Proximal to the active site serine) and a D-pocket (distal to the active site serine. Nevertheless, we consider that through the absent of X-ray data of the thrombin complexes with inhibitors containing in its structure fatty acid moiety it will be correct to consider the interaction of our new inhibitor with a whole hydrophobic cage without its division on pockets, as it is shown in Fig. (4).
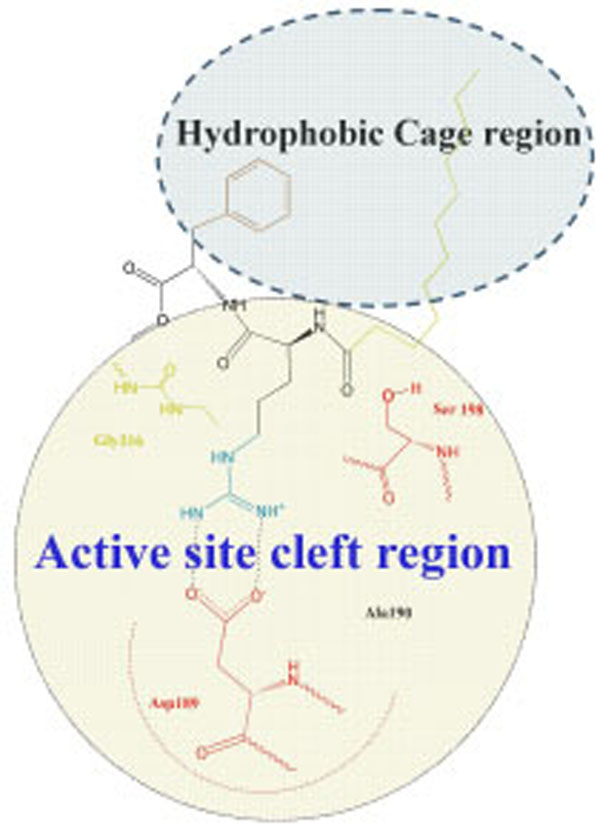
Schematic representation of interaction of inhibitor Laur-DArg-DPhe-OMe with a whole hydrophobic cage without its subdivision on pockets.
This supposition is confirmed by the enough low inhibition constants of both LAE (Кі=2,0 µМ) and Laur-D-Arg-D-Phe-OMe (Кі=1,7 µМ).
In the structure of the compound Chrom-D-Arg-D-Phe-OMe the chromone nucleus is remote from α-amino group of D-arginine by two methylene groups that it results in more conformation mobility of the peptide as a whole and makes possible an interaction of the chromone nucleus with hydrophobic site of the secondary binding site of thrombin credible. Since the secondary binding center of thrombin is in direct proximity to the primary binding site, the competitive character of inhibition of these compounds, by experimental data of kinetic curves, complies with the suppositions made.
CONCLUSIONS
This study have demonstrated unexpectedly high inhibition activity of compounds containing the lauric acid residue despite the traditional stereotype of structures of synthetic thrombin inhibitors containing aromatic substituents only. The discovery of sufficient effect of fatty acid residue on the potency of thrombin inhibitors opens up new possibilities to design new anticoagulants with the fatty acids moiety in future.
ACKNOWLEDGEMENT
We are grateful to colleagues from Grupo LAMIRSA(Laboratorios Miret, S.A and Venta de Especialidades Químicas, S.A.); Roger Segret, Sergi Figueras and Joan Seguer for participation in discussion of this material.
REFERENCES
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]

