All published articles of this journal are available on ScienceDirect.

Repositioning Adequate Antibiotics to Treat/Cure the Coronavirus Disease 2019 (COVID-19): Current Treatments and Future Directions
Authors Info & Affiliations
Abstract
Aims:
Rational use of antibiotics against the betacoronavirus SARS-CoV-2 responsible for the COVID-19 pandemic.
Objective:
Repositioning and repurposing adequate antibiotics to cure the Coronavirus Disease 2019 (COVID-19).
Background:
It is widely accepted that viral infections such as the SARS-CoV-2 cannot be cured by antibiotics, whereas bacterial infections can. It is because the SARS-CoV-2 virus has no protein synthesis machinery (usually targeted by antibiotics) to produce from its RNA genome, the viral proteins and enzymes essential for its replication and/or for the assembly of viral particles. However, the antibiotics must be capable of inhibiting the ribosomes of the protein synthesis machinery of the SARS-CoV-2-infected human host cells, in order to prevent them from synthesizing new proteins that they do not need, but are needed for the virus to spread. Unfortunately, the only antibiotic capable of selectively inhibiting the human 80S ribosomes, namely cycloheximide, was found to be a poisonous drug for the mammals. Therefore, the only possibility is to search for the antibiotics that are capable of inhibiting both bacterial and eukaryal ribosomes, in order to prevent at the same time the ribosomes of the infected human host cells from synthesizing the proteins and enzymes for the SARS-CoV-2 virus, and those of the eventual opportunistic pathogenic bacteria from developing pneumonia.
Methods:
First, we have used a molecular modeling study involving the tools of the semi-empirical quantum mechanics PM3 method to study the interaction between the cation Zn++ and all the molecules considered as zinc transporters in this report. By this approach, the niche in which Zn++ is located was determined. Such an interaction serves as a shuttle and allows zinc cation to invade endocellular structures in the SARS-CoV-2-infected human host cells. Second, we have measured the poly (U)-dependent poly (Phe) synthesis activity of human 80S ribosomes in the presence of increasing concentrations of four antibiotics of the class of the macrolides, namely erythromycin, azithromycin, clarithromycin and telithromycin. This experiment led us to determine for each macrolide, the half-inhibitory concentration (IC50) that is the concentration of antibiotic corresponding to 50% inhibition of the activity of the human 80S ribosomes. Finally, we have analyzed previously published data from the group of Nierhaus (Berlin) on the competition between the incoming aminoacyl-tRNA and the antibiotic tetracycline for the binding to the ribosomal A-site on the E. coli 70S or rabbit liver 80S ribosomes. This led to the conclusion by the authors that tetracycline most likely binds to corresponding sites in 70S and 80S ribosomes with comparable affinity.
Results:
We propose to reposition the macrolides (azithromycin or erythromycin or others) and tetracyclines for the treatment of COVID-19 patients, on account of the following data gathered in this report. First, these antibiotics are already currently successfully used in medicine in humans and animals. Second, the binding sites of these antibiotics at the upper part of the protein exit tunnel (for the macrolides) and the ribosomal A-site (for tetracyclines) are universally conserved features of the ribosomes in all kingdoms of life. So, these classes of antibiotics are expected to bind to all kinds of ribosomes, the 70S as well as the 80S type, with comparable affinity. Therefore, they are capable of preventing at the same time the ribosomes of the infected human host cells from synthesizing the proteins and enzymes for the SARS-CoV-2 virus, and those of the eventual opportunistic pathogenic bacteria from developing pneumonia. Third, the efficacy assessment of these antibiotics in clinical application consisted of comparing their affinity constants of binding to the human ribosomes with their blood concentration.
For example, in the case of azithromycin, the amount of antibiotic administered to COVID-19 patients was 100 μg/ml of circulating blood, which is 43 times superior to the half-inhibitory concentration (IC50 or KIa of 2.3 μg/ml), the concentration of azithromycin corresponding to 50% inhibition of the activity of the human 80S ribosomes. Fourth, zinc cations were previously shown to be a strong antiviral agent, while all the macrolides and tetracyclines that we propose for repurposing or repositioning to cure the COVID-19 are shown in the present report to form Zn++-antibiotic complex and behave as efficient zinc transporters into the SARS-CoV-2-infected host cells.
Conclusion:
The macrolides (azithromycin or erythromycin or others) and tetracyclines selected for repositioning and repurposing to cure COVID-19 are candidates as specific and effective therapeutic drugs available for the coronavirus disease. We propose to combat the current COVID-19 pandemic with azithromycin or erythromycin (or equivalent) alone or in combination with tetracycline (or equivalent) in the presence of Zn++(SO4--). Taking into account the fact that azithromycin had been shown to be effective in treating viral infections such as papillomaviruses in humans and dogs, we conclude that the statement “no antibiotic for viral infections !” is not relevant for all the clinically approved classes of antibiotics, because selective antibiotics such as the universal antibiotics described in the present report are capable of exhibiting antiviral activities through specific interactions with the human 80S ribosomes of infected host cells. As a conclusion, even though the clinical and experimental data presented here do not suggest virucidal activity of azithromycin-zinc or tetracycline-zinc complexes, they do indicate that when administered simultaneously at the onset of first signs of COVID-19, the most common symptoms being fatigue, fever, dry cough, headache, sore throat, muscle pain or shortness of breath, azithromycin (or tetracycline) and zinc cations are capable of inhibiting ribosomal activity of SARS-CoV-2-infected human cells. This results in blocking protein and enzyme synthesis vital for viral RNA replication and for assembly of viral particles. Early treatment allows both reductions of viremia as well as stabilizing symptoms. The major advantage of this therapeutic strategy is avoiding prolonged clinical COVID-19 disease with contingent worsening of illness and subsequent need for intensive care. Prolonged COVID-19 illness is the major downfall of the present pandemic, returning to normal being long, difficult, and sometimes impossible.
1. INTRODUCTION
The Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) is responsible for the so-called coronavirus disease 2019 (COVID-19) [1-3]. Officially, the first identification of the disease occurred in December 2019 in Wuhan, the capital of China's Hubei province [1-3]. The most significant signs and symptoms include fatigue, muscle pain, fever, upper respiratory symptoms such as sneezing, runny nose, sore throat and cough, and shortness of breath, abdominal pain, etc, stemming from the SARS-CoV-2 infection of the respiratory tract [1-3]. However, the coronavirus can also pass into the brain, causing neurological symptoms such as headaches, loss of smell and taste, etc [3]. Other symptoms include gastrointestinal symptoms such as nausea, vomiting and diarrhea [1, 3]. It should be noted that all the people who are infected do not develop symptoms [1, 3]. The time from exposure to onset of symptoms is about five days but may range from two to fourteen days [1, 3]. It is interesting to note that the majority of cases result in mild symptoms, while some evolve to pneumonia and multi-organ failure [3]. Primary spreading of the virus between people occurs during close contact or via small droplets projected by coughing, sneezing, or talking [1, 3]. It is generally thought that, when the droplets are projected by breathing out, they fall onto the ground or on surfaces and are not infectious over long distances [3]. At present, the SARS-CoV-2 has caused hundreds of thousands of deaths worldwide. However, no clinically approved vaccines or specific therapeutic drugs are available for COVID19 at present. In the context of this unprecedented SARS-CoV-2-dependent COVID-19 pandemic, drug repositioning and repurposing appears to be the most promising approach. Re- positioning consists in deploying existing licensed drugs such as antibiotics for newer indications such as the current Coronavirus Disease 2019 (COVID-19). Actually, this approach represents the quickest way to provide clinically effective therapies to those who have contracted this highly contagious disease, with the hope that a vaccine will be developed as soon as possible. In this regard, the most advanced therapy throughout the world so far consists in a tritherapy associating azithromycin, hydroxychloroquine and zinc (II) sulfate, all three active principles being administered together at the onset of COVID-19, upon the first manifestation among the most common symptoms cited above (reference Raoult). Note however, that, in the meantime, a controversial issue [4] had emerged about the role of hydroxychloroquine in the afore- mentioned tritherapy. Other treatments include convalescent plasma, remdesivir, etc. In the present report, prior to re- positioning existing therapeutics to combat the COVID-19, we have evaluated the potentialities of azithromycin, hydroxy- chloroquine and zinc (II) sulfate for the treatment of this disease.
2. MATERIALS AND METHODS
2.1. Materials
Poly(U) and tRNA from E. coli were from Sigma-Aldrich and L-[14C(U)]Phenylalanine (18 GBq/mmol) or L-[3H] Pheny- lalanine (4740 GBq/mmol) from Perkin Elmer. tRNAPhe was aminoacylated using [14C(U)]Phenylalanine or [3H] Phenyla- lanine with an excess amount of partially purified pheny- lalanyl-tRNA synthetase from E. coli [5, 6]. [3H]Phe-tRNAPhe (128 GBq/mol) was acetylated as previously described [5, 6]. All antibiotics were generous gifts of Prof. K. Nierhaus.
2.2. Methods
2.2.1. Molecular Modeling
All studied structures were built starting from a 2-D-ChemDraw structure. Each .cdx file was converted using Chem3D to generate a .mol file. Each 3D-structure was built starting from the molfile obtained from the data bank of the European Bioinformatics Institute (EMBL-EBI). MDL Molfile is indeed a file format containing information about the atoms, bonds, connectivity and coordinates of a molecule and this format is readily implented in the HyperChem package. The resulting 3D-structure was extensively energy-minimized first using molecular mechanics OPLS (Optimized Potentials for Liquid Simulations) force field and then subjected to a molecular dynamics trajectory (target temperature 310 K). Using semi-empirical quantum mechanics PM3, resulting structures were then energy-minimized down to a gradient of 0.01 (~5000 iterations). Indeed, PM3 (for Parametric Method 3) derived from AM1 (Austin Model 1) is a classical semi-empirical method for the quantum calculation of molecular electronic structure in computational chemistry. It is based on the “Neglect of Differential Diatomic Overlap integral” approximation. For this purpose, we employed the handy Polak-Ribiere Nonlinear conjugate gradient method. The resulting low-energy structures were then empirically exposed to a zinc(II) cation probing different loci to search for potential interactions with Lewis base sites. Subsequently to all this, after PM3 calculations, due to excessively huge time-consuming computation times, a limited number of calculations (essentially energy assessments and energy minimization processes) were also carried out using “Ab Initio” and “Density-Functional Theory” (DFT) Quantum mechanics at 6-31G levels to take into consideration zinc 3d orbitals (zinc electronic configuration [Ar] 4s2 3d10).
2.2.2. Poly(U)-dependent Poly(Phe) Synthesis Activity
Poly(Phe) synthesis was determined as incorporation of L-[3H]phenylalanine into material insoluble in hot trichloroacetic acid, as described in a study [5, 6]. The final reaction mixture (50 μl) contained Tris-HCl (pH 7.5, 40 mM), MgCl2 (7 mM), NH4Cl (80 mM), dithiothreitol (DTT, 1 mM), ATP (1 mM), phosphoenol pyruvate (1 mM), GTP (0.5 mM), pyruvate kinase (50 μg/ml), tRNAPhe [2.5 μM, first charged during a 30 min incubation at 30°C with a threefold excess of L-[3H]phenyla- lanine (37 GBq/mmol) and a saturating amount of partially purified phenylalanyl-tRNA synthetase], EF-1α (0.5 μM), EF-1β (0.15 μM), and EF-2 (0.35 μM). The reaction was started with a mixture (14 μl) containing human 80S ribosomes (14 pmol) and poly(U) (4.2 μg), with final concentrations of 0.28 μM ribosomes. During incubation at 37°C, samples (15 μl) were withdrawn at indicated times and spotted on glass fiber filters, and hot trichloroacetic-acid-insoluble radioactivity was determined.
3. RESULTS AND DISCUSSION
3.1. Structure of SARS-CoV-2
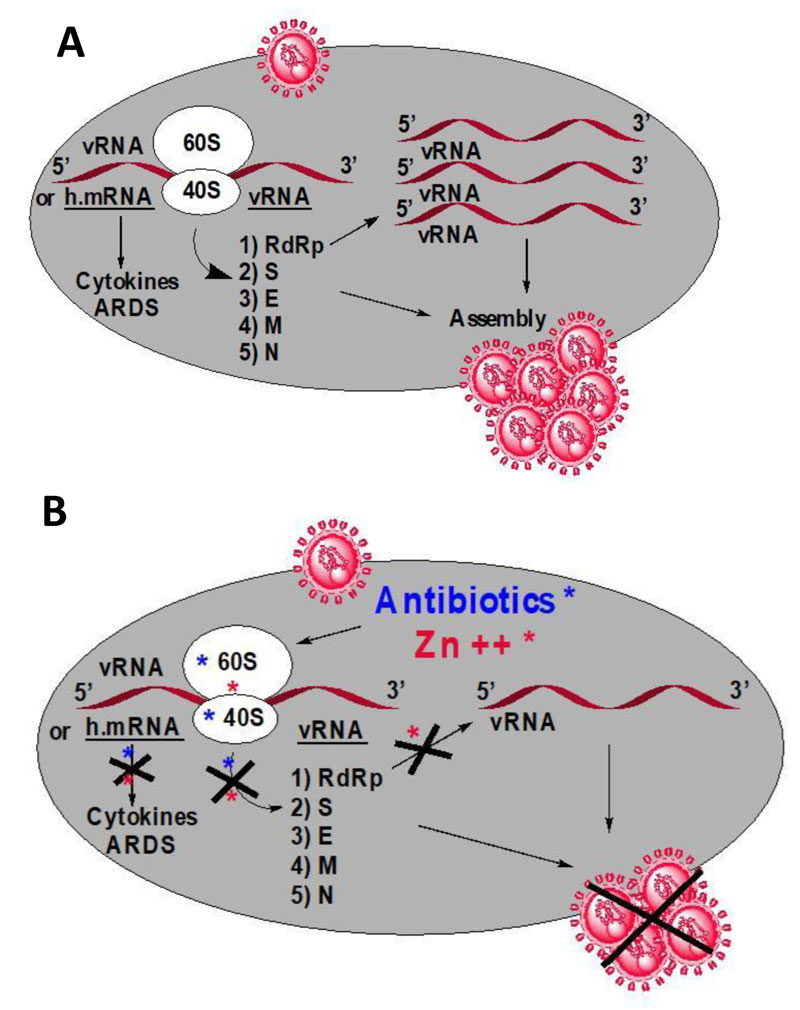
SARS-CoV-2 is an enveloped, positive-sense (+RNA), single-stranded RNA virus which enters the host cell by binding to the Angiotensin-Converting Enzyme 2 (ACE2) receptor [3]. The SARS-CoV-2 genome (30 kb in size) encodes a large polyprotein that is cleaved by proteases to generate 15/16 proteins, 4 structural proteins and 5 accessory proteins (ORF3a, ORF6, ORF7, ORF8 and ORF9) [3]. The four structural proteins include the Spike (S) surface glycoprotein, the Membrane (M) protein, the Envelope (E) protein and the Nucleocapsid (N) protein, all of which are essential for SARS-CoV-2 assembly and infection [3]. The arrangement of the viral genome is in the order of 5′RNA replicase (ORF1a/b)-structural proteins [Spike (S)-Envelope (E)-Membrane (M)-Nucleocapsid (N)]-3′ [3]. The spike surface glycoprotein binds to the ACE2 receptor to permit the entry of the virus into host cells. It can be cleaved by host proteases into an Nterminal S1 fragment and a membranebound Cterminal S2 fragment [3]. The events that follow the entry of the virus into the host cell are dependent on a few proteases belonging to the host cell, such as cathepsins, human airway trypsin-like protease or transmembrane protease serine 2 [3]. The RNA replicase common to all RNA-containing viruses without any DNA stage is an RNA-dependent RNA polymerase (RdRp), which catalyzes the synthesis of the RNA strand complementary to the viral RNA genome as a template [3]. Thus, RdRp is the core enzyme of the viral RNA-synthesizing machinery responsible for the replicative cycle of SARS-CoV-2, and as such, represents a key target for antiviral drug development [3] (Fig. 1A). At this stage, the drug targets for therapeutically useful small molecules with the goal of curing the coronavirus disease 2019 (COVID-19) are: the human angiotensin-converting enzyme 2 (hACE2), the papain-like protease, the RNA-dependent RNA polymerase (RdRp) (Fig. 1A), the helicase, the human dipeptidyl peptidase IV, the Receptor-Binding Domain (RBD), the protease cathepsin L and the type II transmembrane serine protease [3].
3.2. Current Treatments of the Coronavirus Disease 2019 (COVID-19)
Examples of therapeutics that have been used to cure the Coronavirus Disease 2019 (COVID-19) include Remdesivir, Ribavirin and Tocilizumab [7-9]. As discussed above, RdRp represents a key target for antiviral drug development [3] (Fig. 1A), because it is the core enzyme of the viral RNA-synthesizing machinery responsible for the replicative cycle of SARS-CoV-2. For example, Remdesivir [7] is an adenosine nucleoside triphosphate analogue that inhibits RdRp, thereby causing a decrease in viral RNA production. Similarly, the purine analogue Ribavirin [8] is a synthetic antiviral ribonucleoside with activity against a broad spectrum of RNA viruses [3]. In particular, it had been previously demonstrated that Ribavirin triphosphate is an inhibitor of the viral RdRp [8]. In contrast to the two former antiviral drugs that target RdRp, Tocilizumab [9] is used for selective blockade of interleukine-6 (IL-6) receptor. The inflammatory cytokine IL-6 is released by large numbers of activated white blood cells, leading to the COVID-19-related cytokine release storms [10] and Fig. (1A) triggered by SARS-CoV-2 infection, responsible for countless deaths [10]. However, the most widely used treatment throughout the world at the beginning of the pandemic was a tritherapy that combined azithromycin, hydroxychloroquine and zinc (II) sulfate [11]. Evaluation of the potentialities of each of these active principles in the present report consists in describing their role in the cure/treatment of the COVID-19 disease.
3.3. To Combat the Coronavirus SARS-CoV-2, all We Need is Zinc Cation
As a starting point, we assume that Zn++ can be considered as a strong antiviral agent in COVID-19 treatment (Fig. 1B) [12-15]. First, it had been previously demonstrated that Zn++ inhibits coronavirus RNA-dependent RNA polymerase (RdRp), the RNA replicase which catalyzes the synthesis of the RNA strand complementary to the viral RNA genome [3, 12] and (Fig. 1B). Inhibition of the viral RdRp by Zn++ results in preventing the replication of the viral RNA genome of SARS-CoV-2 [12] and (Fig. 1B). Second, Zn++ had been previously shown to inhibit translation initiation [13], suggesting that not only the zinc ion is an effective inhibitor of RdRp, but it is also capable of preventing the synthesis of this RNA-synthesizing machinery responsible for the replicative cycle of SARS-CoV-2 (Fig. 1B). Third, zinc was previously shown to impair the replication of RNA viruses, by interfering with the correct proteolytic processing of viral polyproteins [14]. Fourth, it was previously demonstrated that zinc is capable of improving the antiviral immunity through up-regulation of interferon α production thus increasing antiviral and anti-inflammatory activities [14]. Altogether, these observations suggest that all small-molecule inhibitors capable of serving as zinc transporters into SARS-CoV-2-infected host cells would exhibit protective and therapeutic effects against COVID-19.
3.4. Chloroquine or not Chloroquine?
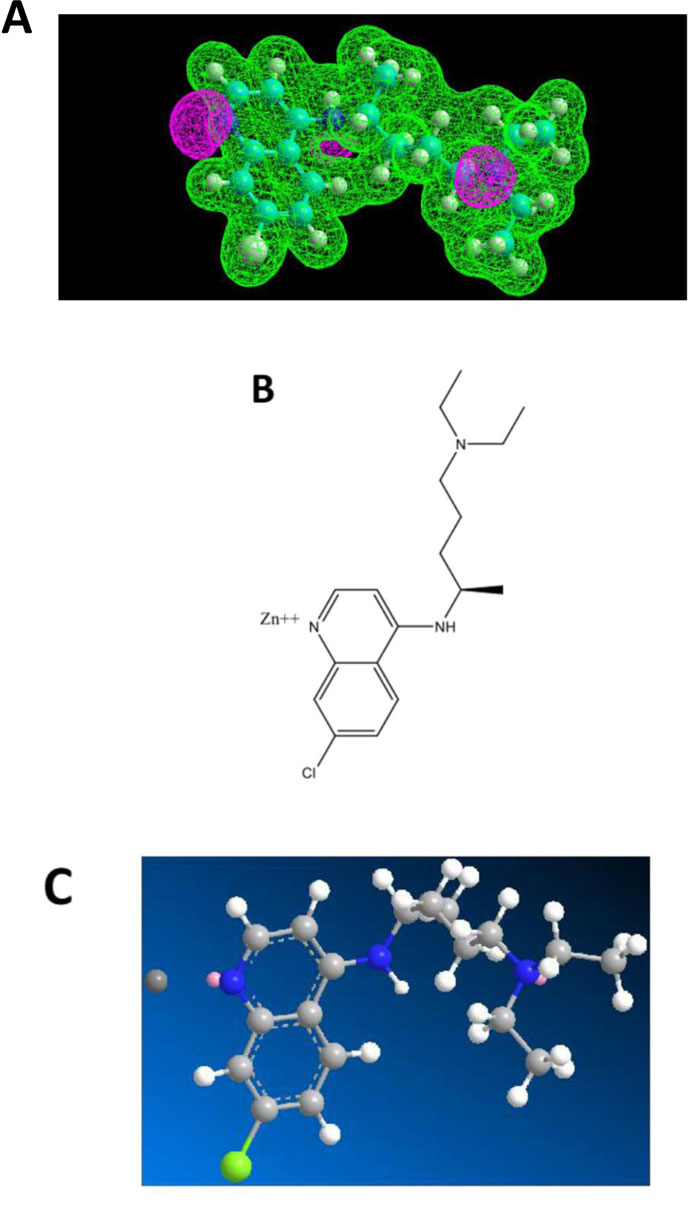
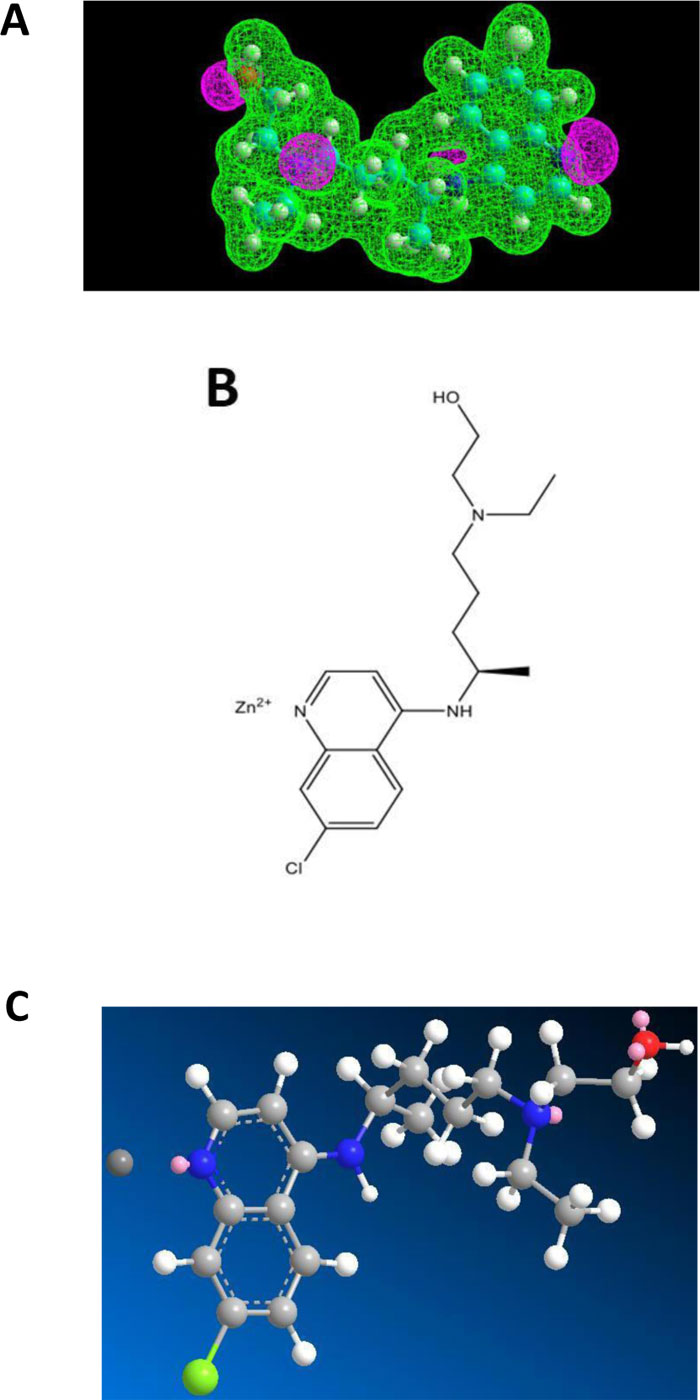
Chloroquine (CQ) and Hydroxychloroquine (HCQ) belong to the class of 4-aminoquinoleine, a most druggable privileged template. These two molecules have a long past in the therapeutic armamentum as antimalarial drugs. However, due to the emergence of drug resistance, they are no longer used in this indication. HCQ, however, found a niche in the treatment of rheumatoid arthritis. More recently, they came back on the forefront in the context of COVID-19 management, more specifically in the tritherapy (HCQ - azythromycin - zinc sulfate) proposed by Prof. D. Raoult of Marseille, France [11]. In general, this cure was badly received internationally. It should be pointed out that Raoult emphasized that his treatment was to be implemented early in the development of COVID-19 [11]. Most opponents quickly jumped to negative conclusions after application of CQ and HCQ alone to patients. Among the criticisms, in addition to its alleged contextual inefficacy, they mentioned problems of toxicity for HCQ. How is it possible that a drug used safely for decades for arthritis and listed as an essential drug by the WHO all of a sudden becomes toxic at the dosages recommended by Raoult, which are essentially those applied for rheumatoid arthritis? As a matter of fact, used alone, HCQ doesn’t have a chance to be active against COVID-19. It is the combination therapy that acts efficiently and where the zinc cation is the main actor. The purpose of the present report is to disclose the inner mechanisms behind the successful tritherapy of Raoult and coworkers [11]. To this end, we have conducted a biomolecular study in order to depict a potential interaction between Zn++ and CQ or HCQ. The Electrostatic Potentials contour of CQ showed three nitrogen atoms, one of which belonging to a tertiary amine group representing a potential interaction site for the zinc(II) cation (Fig.2A). In addition, HCQ contains one hydroxyl group at one extremity (Fig.3A). The model obtained accounted on an only one-point attachment (i.e. the tertiary amine group present in both CQ and HCQ) (Figs. 2A and 3A), while zinc(II) cation is well-known to have a co-ordinance of four ligands. We, therefore, conclude that such a weak interaction (Figs. 2B, 2C, 3B and 3C) might account for a rather poor affinity of CQ and HCQ for Zn++, and would suggest at the same time that these molecules might not serve as an efficient shuttle allowing zinc cation to easily invade endocellular structures. It remains that, at the early stage of the disease, even a low amount of Zn++ might be sufficient to exert protective and therapeutic effects against COVID-19. Another role of CQ and HCQ is related to the prevention of the viral infection. Briefly, for entry into host cells, the SARS-CoV-2 virus binds to the ACE2 receptor surface molecules by means of the viral spike surface glycoprotein.
The events that follow the entry of the virus into the host cell are dependent on a few proteases belonging to the host cell, such as cathepsins. These acidic pH dependent proteases require endosomal/lysosomal acidification. Interestingly, CQ and HCQ are weak bases which are capable of increasing the pH [16] of acidic cytoplasmic vesicles such as endosomes, lysosomes, or Golgi vesicles by diffusing spontaneously and rapidly across the membranes of cells and organelles. Therefore, the inhibition of endosomal acidification by CQ and HCQ is expected to inhibit viral entry, replication and infection. However, the in vivo antiviral effects of CQ or HCQ on the SARS-CoV-2 virus have not been proven so far.
3.5. The Role of Azithromycin in the Treatment of the COVID-19 Disease
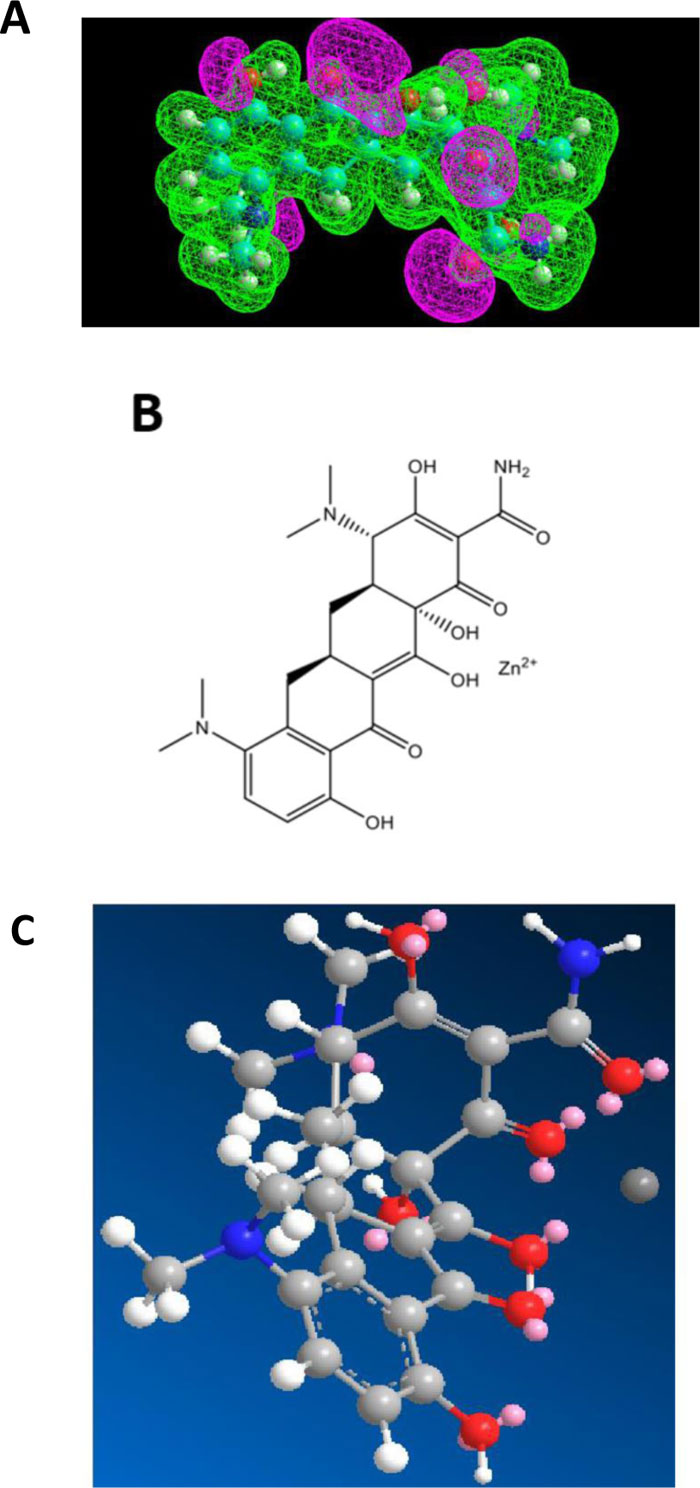
The first role of azithromycin (an antibiotic of the macrolides class) in the treatment of the COVID-19 disease is related to the activity of Zn++ as a strong antiviral against COVID-19. In fact, we have demonstrated recently that azithromycin is capable of forming a Zn++-antibiotic complex that serves as zinc transporter in SARS-CoV-2-infected host cells treated with this antibiotic in the presence of zinc cations [15]. Fig. (4A) shows that azithromycin exhibits an electron-rich pocket, which represents a potential interaction site for a strong Lewis acid such as the zinc(II) cation. Accordingly, the docking of Zn++ on azithromycin showed the zinc cation bound in the middle of this pocket (Figs. 4B & C). These results are likely to reflect a strong binding affinity of Zn++ for azithro- mycin, and suggest at the same time that this antibiotic is a better zinc transporter than chloroquine or hydroxychloroquine. In this view, the administration of the Raoult’s therapy without chloroquine or hydroxychloroquine might have similar positive effects without adverse side-effects of these aminoquinoleine molecules.
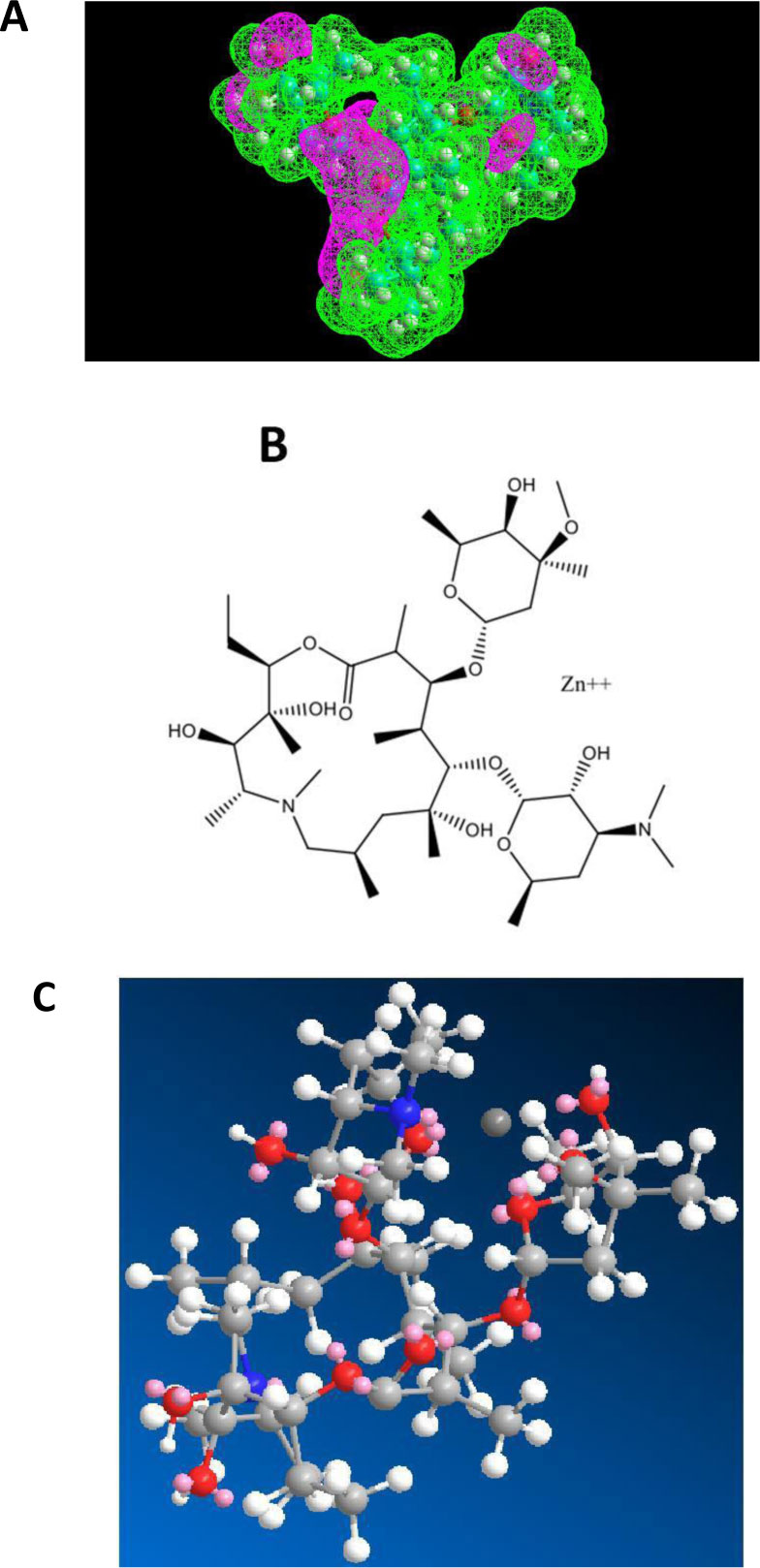
As shown in Figs. (5A-C), erythromycin is also capable of forming a Zn++-antibiotic complex.
The fact that both azithromycin and erythromycin are equally capable of forming a Zn++-antibiotic complex makes it possible to choose among the family of the macrolides, the antibiotic which has less side-effects for the treatment of the coronavirus disease. It is interesting to note that we have recently demonstrated the role of calcitriol (the active form of vitamin D3) as a zinc transporter, in accordance with the fact that there is a solid correlation between vitamin D3 deficiency and low serum levels of zinc (personal communication of J. Poupaert). Accordingly, we had proposed that the Calcitriol:Zinc (II) Cation complex might represent a useful tool for the prophylaxis of SARS-CoV-2. Another specific role of azithromycin as an antibiotic in protein synthesis on the ribosome of SARS-CoV-2-infected human host-cells will be discussed in the next section.
3.6. Protein Synthesis on the Ribosome
Protein biosynthesis is carried out by the ribosome in all kingdoms of life. The ribosome is a ribonucleoprotein particle composed of two subunits of unequal size (Figs.6A & B) [17]. The large subunit (50S in eubacteria and archaeabacteria, and 60S in eukaryotes) contains the active site, the Peptidyl Transferase Center (PTC), which catalyzes the formation of peptide bonds in the growing polypeptide, while the small subunit (30S in bacteria and 40S in eukaryotes) carries the decoding site where an mRNA codon is recognized by the anticodon of its cognate aminoacyl-tRNA (aa-tRNA) [17].
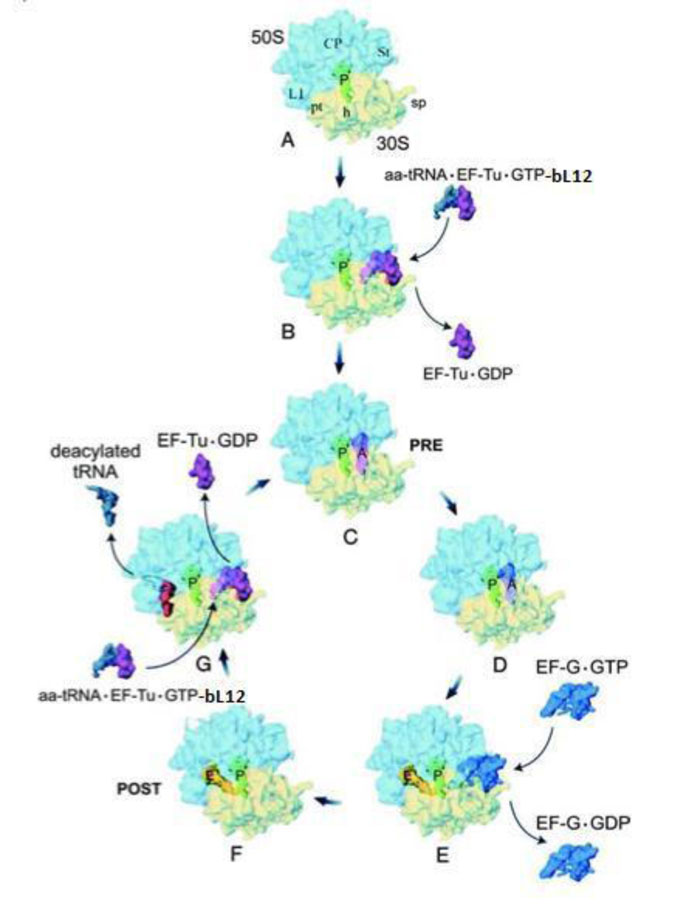
Translation is a cyclic reaction, which can be divided into four sequential steps: initiation, elongation, termination and recycling (Fig.7) [17]. The elongation cycle is the heart of protein synthesis whereby each “heart beat” of the ribosome lengthens the nascent polypeptide chain with one amino acid. The elongation cycle can be divided into the following three basic reactions: (i) Binding of the incoming aminoacyl-tRNA (aa-tRNA) to the ribosomal A-site. This step can be further subdivided into (a) the decoding reaction, which consists in the codon–anticodon interactions (low-affinity interaction) between the aa-tRNA and the ribosome and (b) the accommodation reaction, where high-affinity binding of the aa-tRNA to the A-site leads to the fitting of the aminoacyl moiety into the Peptidyl Transferase Center (PTC), the active site located on the large subunit. (ii) Peptide-bond formation, where the nascent polypeptide chain is transferred from the P-site tRNA onto the aminoacyl moiety of the A-site-bound aa-tRNA. This leaves a deacylated (or uncharged) tRNA at the P-site and a newly formed peptidyl-tRNA at the A-site (with the nascent chain lengthened by one amino acid). (iii) Translocation involves the movement of mRNA•tRNA on the ribosome by one codon length so that the deacylated tRNA moves to the E-site and the peptidyl-tRNA to the P-site, thus leaving free the A-site for the next incoming aminoacyl-tRNA. Translocation shifts the ribosome from a Pre-translocational (PRE) state to a Post-translocational state (POST) (Fig. 7). Ongoing of the translating ribosome through these various stages of the elongation cycle is catalyzed by protein factors called elongation factors, specifically elongation factor G (EF-G) and elongation factor Tu (EF-Tu) in bacteria [17]. A comprehensive overview of the elongation cycle is shown in Fig. (7). Before translocation (PRE in Fig. 7) the tRNA molecules are on the A- and P-sites and, following translocation (POST as shown in Fig. 7), the tRNA molecules are on the P- and E-sites. During translation, the nascent peptide exits from the ribosome through a tunnel that extends from the PTC up to the bottom (cytoplasmic side) of the large subunit [17].
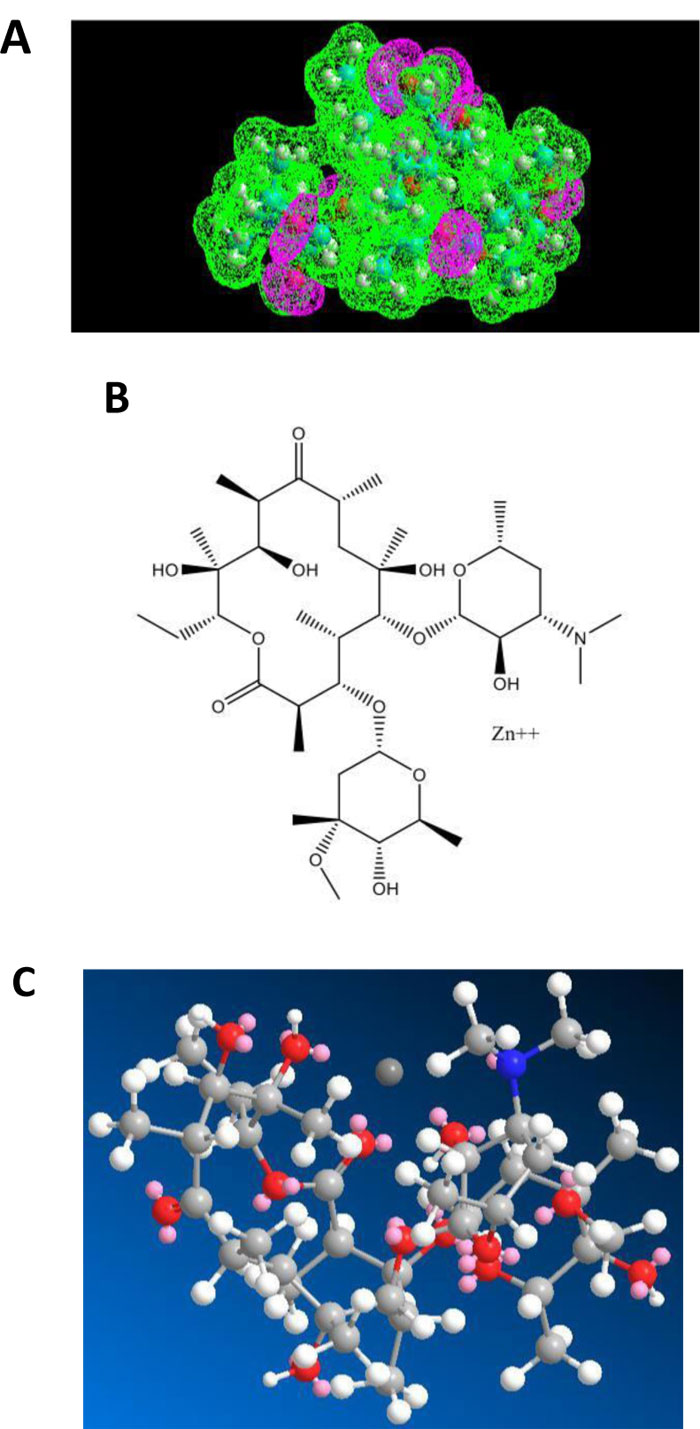
3.7. Another Role of Azithromycin in the Treatment of the COVID-19 Disease
As shown in Fig. (1), the ribosome of SARS-CoV-2-infected human host-cells is the primary target for the antibiotics because, with the exception of the RNA genome, the SARS-CoV-2 virus contains neither any major molecular material nor any protein synthesis machinery. Therefore, whether they belong to the SARS-CoV-2 virus or to the infected human host cell, all the enzymatic or non enzymatic proteins present in the infected cells are synthesized by the protein synthesis machinery built up with the human ribosomes (Fig. 1). In other words, the virus forces the host cell to synthesize new proteins that the cell does not need, but are needed for the virus to spread (Fig. 1). Thus, in order to prevent the ribosomes of the infected human host cells from synthesizing the viral proteins and enzymes, selective inhibition of the human 80S ribosomes with eukaryote-specific antibiotics is needed. This is a task for cycloheximide (Table1), the most common antibiotic specific for the ribosomes of eukaryotic cells that have proven to be a strong inhibitor of translation and a potent antitumor drug. Unfortunately, cycloheximide was found to be a poisonous drug for mammalians. Therefore, we have selected the antibiotics that are capable of inhibiting both eukaryal and bacterial ribosomes, in order to prevent at the same time the ribosomes of the infected host cells from synthesizing the viral proteins and enzymes, and those of the eventual opportunistic pathogenic bacteria from developing pneumonia. As shown in Table 1, there are only two classes of antibiotics that fulfill this criteria. These are macrolides (including azithromycin and erythromycin) and tetracyclines. It is interesting to note that the majority of researchers, physicians, and healthcare professio- nals have mistakenly prohibited the use of antibiotics, because viral infections cannot be cured by antibiotics whereas bacterial infections can. However, regarding azithromycin that is currently widely used worldwide to cure COVID-19, they all have declared that this antibiotic was expected to prevent opportunistic pathogenic bacteria present in the respiratory tract from developing pneumonia in parallel with the SARS-CoV-2 infection [10]. In other words, it is assumed that opportunistic pathogens are likely to represent primary targets for azithromycin or any other antibiotic for the treatment of COVID-19 pneumonia. However, it is unlikely that the same opportunistic pathogenic bacteria responsible for a surin- fection-related pneumonia and sensitive to azithromycin would be widespread across the whole planet along with the SARS-CoV-2 virus responsible for the COVID-19 pandemic. In conclusion, it is most probable that the primary targets for azithromycin are not opportunistic pathogens.
| Inhibitors of the small ribosomal subunit | ||
|---|---|---|
| Antibiotics | Class | |
| B/E | Tetracycline | Tetracycline |
| B | Streptomycin | Aminoglycoside |
| B | Paramomycin | Aminoglycoside |
| E | Geneticin G418 | Aminoglycoside |
| Inhibitors of the large ribosomal subunit | ||
| B/E | Sparsomycin | Glutarimide |
| E | Cycloheximide | Glutarimide |
| B/E | Erythromycin | Macrolide |
| B/E | Azithromycin | Macrolide |
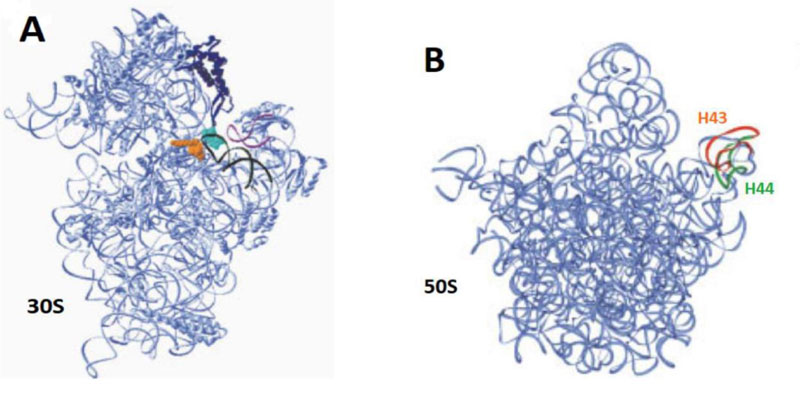
3.8. Azithromycin and Tetracycline are Suitable to Inhibit the Human 80S Ribosome of SARS-CoV-2-Infected Host-cells because they Target Evolutionarily Conserved Structural Features in all Kingdoms of Life
Considering the essential function of ribosomal protein synthesis in all kingdoms of life, it is not surprising that this process is a target for many small-molecule inhibitors of translation, the majority of which affect the translation elongation cycle. Four major binding sites for ribosomal inhibitors can be distinguished: (i) The decoding center on the small subunit, (ii) The Peptidyl Transferase Center (PTC), (iii) The exit tunnel and (iv) the GTPase-Associated Center (GAC) on the large subunit. Among these, we assumed that the protein exit tunnel and the A-site where the incoming aminoacyl-tRNA binds represent evolutionarily conserved functional sites in the bacteria and the eukaryotes and are likely to bind similarly azithromycin and tetracycline in all kingdoms of life. The reasoning is the following: (i) Azithromycin belongs to the class of the macrolides (Fig. 8) that represent a large and clinically important class of antibiotics. They bind in the upper part of ribosomal tunnel where they inhibit protein synthesis by interfering with the progression of the nascent peptide (Fig. 9) [18-21]; (ii) Since all proteins in the living cells are built up with the same common 20 or so amino acid residues, it is obvious that the tunnel whereby their 1-dimensional structure exits from the ribosomes should be of comparable size and shape from the top to the bottom in all kingdoms of life; (iii) Similarly, the tetracyclines (Fig. 10) are inhibitors of translation elongation which specifically prevent binding of tRNA to the A-site by interfering with the decoding site of the 30S ribosomal subunit [22-26]. Since the anticodon of all tRNA molecules consists of trinucleotides complementary to the A-site codon on the mRNA, the latter 15 angström-long A-site platform should be universally conserved in all kingdoms of life; (iv) In conclusion, the protein exit tunnel and the A-site platform represent universally conserved functional sites and are likely to bind the macrolides similarly on one hand, and the tetracyclines on the other hand in all kinds of ribosomes, the 70S as well as the 80S type, with comparable affinity. Therefore, these antibiotics are referred to as universal antibiotics; (v) As a matter of fact, it had been previously shown that the relative orientation of the exit site of the nascent polypeptide chain from the tunnel was identical to the ribosomes of eubacteria and eukaryotic cells [27]. Moreover, the alignment of cryo-EM structures from rat liver ribosomes with those of Escherichia coli had proven that the tunnel can be superimposed, consistent with the fact that it is a universally conserved functionally important feature of the ribosome [28]; (vi) ribosomal RNA (rRNA), the main structural and functional component of the ribosome, is characterized by a high degree of evolutionary conservation among the organisms from different origins. In general, many rRNA residues, especially those located in important functional sites (such as the protein exit tunnel or the aminoacyl-tRNA binding site) of the ribosome, that are also the sites of action of many ribosome-targeting antibiotics, are found invariable between the bacteria and the eukaryotes. Therefore, it is generally assumed that these drugs bind in the same, or very similar ways to ribosomes isolated from different bacterial or eukaryotic organisms. However, even though the general location of the macrolide or the tetracycline binding site is the same in ribosomes of different bacterial or eukaryotic organisms, this does not necessarily mean that all the molecular interactions of the drugs with the ribosome, or even the conformation of the ribosome-bound antibiotic is evolutionarily conserved. This observation points to species-specific interactions of the macrolides or the tetracyclines with the ribosome that might account for different binding affinities between antibiotics of the same family toward the functional sites of the ribosomes from different bacterial or eukaryotic organisms. Finally, it was previously reported that the antibiotics that bind to the archaebacterial ribosome could also bind to the eukaryal ribosome as well, because they share several structural features [6, 29, 30].
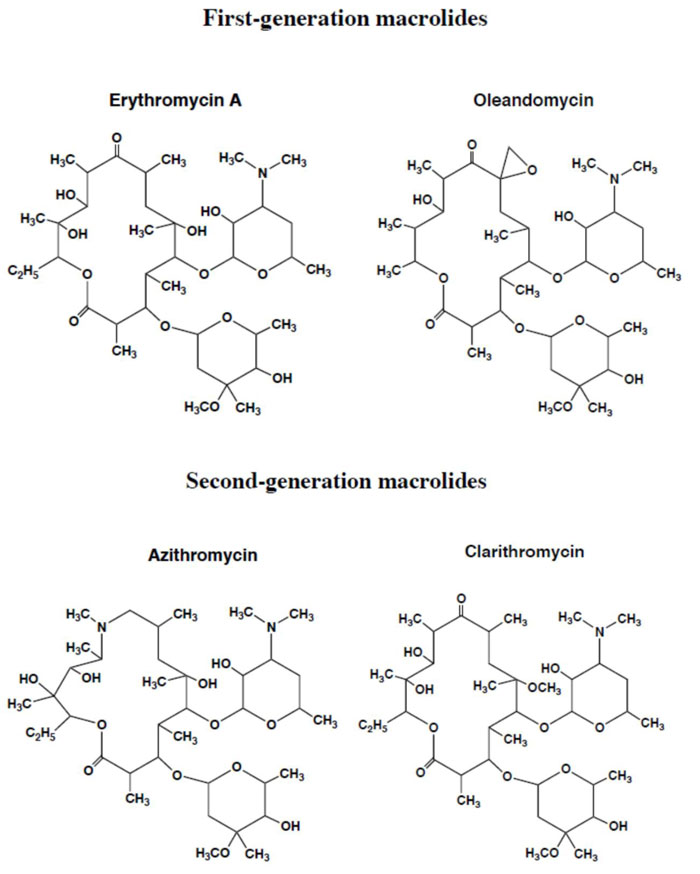
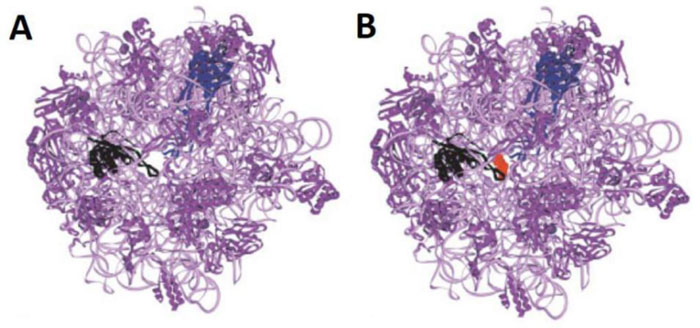
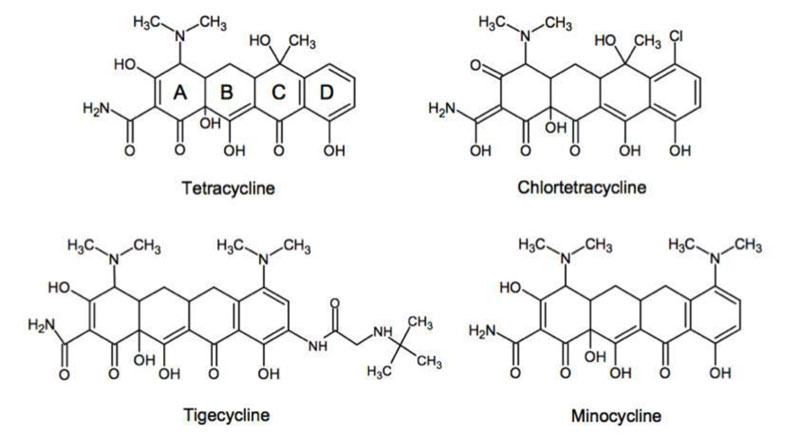
3.9. Rationale for Repositioning Antibiotics to Cure the Coronavirus Disease 2019 (COVID-19)
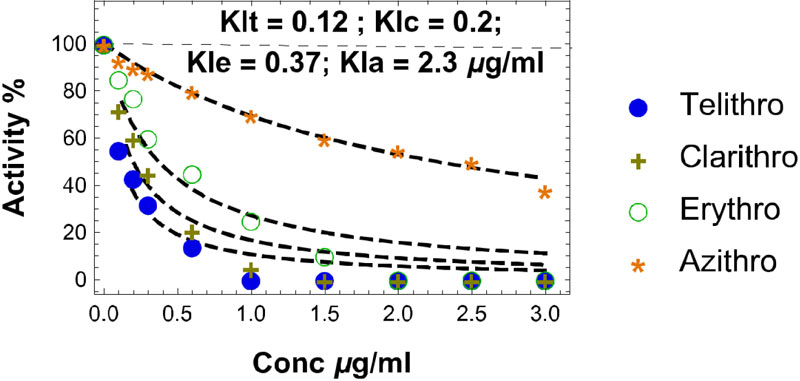
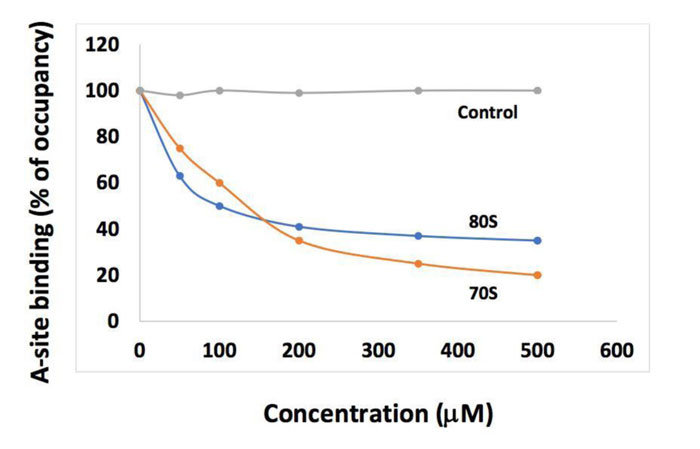
As discussed above, the macrolides and the tetracyclines are capable of inhibiting both eukaryal and bacterial ribosomes, in order to prevent at the same time the ribosomes of the SARS-CoV-2-infected host cells from synthesizing the viral proteins and enzymes (Fig. 1), and those of the eventual opportunistic pathogenic bacteria from developing pneumonia. Fig. (11) shows the schematic functioning of the elongation step of translation in the absence or the presence of azithromycin or tetracycline bound to the prominent functional sites of the bacterial 70S or the human 80S ribosomes. In contrast to the common view of the ribosomal post-crystal-structure era, a quaternary (bL12:aa-tRNA:EF-Tu:GTP) complex (Fig. 7) (instead of the ternary aa-tRNA:EF-Tu:GTP complex) is responsible for the accommodation reaction corresponding to the binding of the aa-tRNA to the A-site and the fitting of its aminoacyl moiety into the Peptidyl Transferase Center (PTC) on the translating bacterial 70S ribosome [31, 32] (Figs. 7 and 11A). In the case of human 80S ribosome, rp bL12 was replaced by the eukaryote-specific rp eL42, while the elongation factor was EF-1α (instead of EF-Tu) (Fig.11B). In Figs. (11C & D), the binding of azithromycin to the entrance of the protein exit tunnel on the 50S or the 60S subunit, inhibits translation elongation by preventing the progression of the nascent peptide chain into the tunnel [18-21]. We have measured the poly(U)-dependent poly(Phe) synthesis activity of human 80S ribosomes in the presence of increasing concentrations of four macrolides. As shown in Fig.(12), the half-inhibitory concentration (IC50) that is the concentration of antibiotic corresponding to 50% inhibition of the activity of the human 80S ribosomes is 0.37, 2.3, 0.2 and 0.12 μg/ml for erythromycin, azithromycin, clarithromycin and telithromycin, respectively. These data would suggest that erythromycin could be more efficient than azithromycin in preventing the ribosomes of the human host cells infected by SARS-CoV-2 from synthesizing the viral proteins and enzymes. However, taking into account that erythromycin had been previously reported to be less stable than azithromycin in the acidic digestive tract [17], both antibiotics should be equally efficient for the inhibition of the ribosomes of the human host cells infected by SARS-CoV-2. Inhibition of translation elongation by tetracycline (Tet) bound to bacterial or eukaryal ribosomes is shown in Figs. (11E & F). In this case, the antibiotic is bound to the A-site where it prevents the binding of the aa-tRNA (Figs. 11E & F) [22-24]. Here, it is the competition between the incoming aminoacyl-tRNA and Tet for the binding to the ribosomal A-site rather than the loss of activity of the ribosome that is to be taken into account [24] and (Fig. 13). The group of Nierhaus in Berlin had previously demonstrated that the A-site binding of AcPhe-tRNA was sensitive to Tet to a comparable extent in the E. coli 70S or rabbit liver 80S ribosomes [24] and (Fig. 13). The authors had concluded that this antibiotic should bind to corresponding sites in 70S and 80S ribosomes with comparable affinity [24]. However, the affinity of [3H]tetracycline binding to free E. coli 70S or rabbit liver 80S ribosomes, as studied by this group had demonstrated that the latter bind tetracycline with an affinity 15 times lower than the former (KD 30 μM and 1-2 μM, respectively) [24]. These data are likely to reflect a preferential inhibition of bacterial rather than eukaryotic ribosomes by tetracycline in vivo. As discussed above, this phenomenon might be due to structural differences of the tetracycline binding site between bacterial and eukaryotic rRNAs. Inhibition of translation elongation by both azithromycin and tetracycline (Tet) bound to bacterial or eukaryal ribosomes is shown in Figs. (11G & H). These antibiotics target both the protein exit tunnel and the A-site with the goal of obtaining a complete inhibition of the ribosomes of the human host cells infected by SARS-CoV-2. Another prominent target for small-molecule inhibitors on the ribosomes of the human host cells infected by SARS-CoV-2 is the newly discovered rp eL42 (formerly L42A or L42AB in yeast, L36a or L36a-like in human, or L44e in archaea) from human 80S ribosomes. We have recently demonstrated that the large subunit ribosomal proteins bL12 and eL42 assist catalysis by correctly positioning the incoming aminoacyl-tRNA in the A-site of 70S or 80S ribosomes of bacteria and eukaryotic cells, respectively [5, 6, 31, 32]. Therefore, we hypothesize that the small-molecule inhibitors, as well as the reactive substrate analogues (R in Figs. 11I & J) that are capable of covalently reacting with the catalytic amino acid side chain of rp eL42 inside the human 80S ribosomes (of the infected host cells), might also be capable of inhibiting protein synthesis on the ribosome.
3.10. Advantages of Repurposing or Repositioning Antibiotics to Cure the Coronavirus Disease 2019 (COVID-19)
The clinical potential and mechanistic targets of the antibiotics in mammalian cells is still not well understood. Antibiotics that block the translation process in mammalian cells have potential as therapeutics for diseases such as cancer, inflammation or COVID-19. Repurposing adequate antibiotics such as macrolides (azithromycin or erythromycin or others) and tetracyclines for these diseases is, therefore, a highly attractive approach. These antibiotics have the following practical advantages: (i) first of all, as discussed above, these antibiotics referred to as universal antibiotics are capable of universally inhibiting the ribosomes in all kingdoms of life, in accordance with their capability to inhibit the translation process in mammalian cells and to represent potential therapeutics for human diseases; (ii) they are widely distributed, inexpensive and not stigmatizing; (iii) some of them have been successfully administered to COVID-19 patients (see below); (iv) they are available and safe and it might be possible to evaluate them in suitable clinical trials whatever the severity of the current pandemic; (v) they had been previously successfully used in medicine (Table 2). For example, azithromycin was found to be effective in humans and dogs with papillomatosis [33] (Table 2). In fact, papillomaviruses induce severe infections in animal and human hosts [33-35]. Papillomas affect the oral mucous membranes and skin of young and older dogs, respectively [33] (Table 2). Papillomavirus is implicated as an aetiological factor in canine oral papillomatosis [33] (Table 2). However, the underlying mechanisms are not completely understood. It was proposed that azithromycin eradicates an unknown organism involved in predisposition to papillomatosis [33]. Another explanation was that azithromycin could help to suppress an autoimmune phenomenon leading to the formation of papillomatosis [33]. Taking into account the mechanism of action of azithromycin in the present report, it becomes obvious that the effectiveness of azithromycin against papillomatosis consists in the inhibition of the ribosomes of the animal host, in order to prevent them from synthesizing the proteins and enzymes of the papillomavirus (Fig. 1). Therefore, on account of these clinical data, we conclude that the statement “no antibiotic for viral infections !” is not relevant for all the clinically approved classes of antibiotics, because selective antibiotics such as the universal antibiotics described in the present report are capable of exhibiting antiviral activities through specific interactions with the human 80S ribosomes of infected host cells. Similarly, several studies had previously demonstrated the so-called non-antibiotic activities of tetracyclines, including anti-inflammatory, antioxidant, anti-apoptotic, and anti-cancer properties [26, 36-39] (Table 2). For example, minocycline has been shown to significantly reduce the secretion of cytokines such as IL-6, IL-2, and TNF-α, suggesting that it has the potential to reduce the risk of disease-associated cytokine release syndrome [10]. In this case, the role of minocycline consists in the inhibition of protein synthesis on the human ribosomes, leading to reduction of the formation of cytokines (Fig. 1). In the case where minocycline is used for the treatment of cancers, its role consists in the inhibition of global protein synthesis on the human ribosomes of cancer cells, in order to slow down cell division and cancer cell proliferation (Table 2). Therefore, the expression “non-antibiotic activities of tetracyclines” is improper. Another example is the tetracycline-class antibiotic doxycycline that is used in the treatment of infections caused by certain parasites such as Plasmodium falciparum, the mosquito-transmitted parasite responsible for the most severe form of human malaria [40] (Table 2). In this case, the Plasmodium ribosome is the target for the antibiotic, which inhibits protein synthesis in the parasite [40], in order to decrease its multiplication (Table 2). Finally, Figs. (14A-C) show that, similar to azithromycin, minocycline or any other tetracycline-class antibiotic is capable of forming a minocycline-Zn++ complex that would act as a zinc transporter. Since, as discussed above, any small-molecule inhibitor capable of serving as zinc transporters into SARS-CoV-2-infected host cells is expected to exhibit protective and therapeutic effects against COVID-19, the fact that all the macrolides and tetracyclines that we propose for repurposing or repositioning to cure the COVID-19 are shown in the present report to form Zn++-antibiotic complex and to inhibit at the same time the human 80S ribosomes making them candidates as specific and effective therapeutic drugs available for COVID-19. As the pandemic was going on worldwide, we have been aware that the majority of the Physicians throughout the world were using azithromycin to cure COVID-19 patients. Fortunately, we met three of them who have become members of our Research Group. On this occasion, they have explained that, on the basis of their medical practitioner habits, and of the general use of azithromycin worldwide, they had previously prescribed azithromycin to 167 COVID-19 patients of 19 to 88 years old, all of which have recovered. In the present report, we have worked together on describing the role of azithromycin in the treatment of COVID-19. In particular, we have been interested in comparing the blood concentration of azithromycin in the treated COVID-19 patients with the affinity constant of the antibiotic, as deduced from the present study (Fig. 12). The prescription used worldwide was 500 mg of azithromycin (trade name Zithromax) per day for 7 days. Considering a mean value of blood volume equal to 5 liters for 500 mg of antibiotic, the calculated blood concentration of azithromycin is 100 μg/ml. This concentration is 43 times superior to the half-inhibitory concentration of azithromycin (IC50 or KIa of 2.3 μg/ml in Fig. 12) that is the concentration of azithromycin corresponding to 50% inhibition of the activity of the human 80S ribosomes. Since a ligand concentration 10 times superior to the value of IC50 is likely to reflect the saturation of a catalyst by this ligand, we can conclude that the blood concentration of azithromycin (100 μg/ml) is largely sufficient to achieve the saturation and complete inhibition of the ribosomes of the SARS-CoV-2-infected human host cells. Considering that the quantity of tetracycline (MW 444 Da) generally prescribed for other indications is 200 mg per day during 7 days, we have calculated a blood concentration of 90 μM for tetracycline in the case where this treatment would be administered to COVID-19 patients. This concentration is 6 times superior to the dissociation constant (KD=15 μM) of binding tetracycline to the mammalian ribosome, suggesting that the translating human 80S ribosomes of the SARS-CoV-2-infected host cells were probably not completely saturated at this blood concentration of tetracycline. In conclusion, we propose to combat the current COVID-19 pandemic with azithromycin or erythromycin (or equivalent) alone or in combination with tetracycline (or equivalent) in the presence of Zn++(SO4--). One should keep in mind the most well-known problem of drug resistance. In consequence, we will test as many antibiotics as possible in the classes of macrolides and tetracyclines, respectively, in order to select the best combinations suitable to cure the COVID-19. Moreover, the immediate benefits against COVID-19 are by far bigger than the long term risks related to the resistance to antibiotics of pathogenic bacteria.
3.11. Cytokines and Inflammation in COVID-19
One major complication of the COVID-19 is related to the cytokine release storms responsible for countless deaths. It is a hyperinflammation response that is likely to be triggered by SARS-CoV-2 infection [10]. It occurs because large numbers of white blood cells are activated and release inflammatory cytokines (including IL-1 and IL-6), which in turn activate yet more white blood cells. Current therapeutic options include steroids, intravenous immunoglobulin, selective blockade of IL-1 with anakinra, and of IL-6 receptor with tocilizumab [9]. Given that cytokines are proteins synthesized by the ribosomes and glycosylated post-translationally by enzymes that are also synthesized by the ribosomes of the infected host cells, the use of individual antibiotics or the combination thereof to cure the COVID-19 is expected to inhibit the ribosomes and prevent cytokines release. Two diseases that are seemingly associated with COVID-19 are the Kawazaki and the Guillain-Barré diseases [41, 42]. The Guillain-Barré Syndrome (GBS) [41] is an autoimmune inflammation of the peripheral nervous system in which the body's immune system mistakenly attacks and damages the peripheral nerves leading to muscle weakness of the feet, the legs, the arms and upper body, affecting both sides equally.
| Disease | Aetiological Factor | Antibiotic | Therapeutic Target |
|---|---|---|---|
| COVID-19 | SARS-CoV-2 This report |
Azithromycin | Human 80S Ribosome |
| Human Papillomavirus |
Papillomatosis M. Atasoy et al., 2014, The J. Dermatol., 31, 1-10. |
Azithromycin | Human 80S Ribosome |
| Dog Papillomatosis |
Papillomavirus BB. Yagci et al., 2008, Vet. Dermatol., 19, 194-198. |
Azithromycin | Dog 80S Ribosome |
| Malaria | Plasmodium falciparum M. Sun et al., 2015, Nucleic Acids Res., 43, 10515-10524. |
Doxycycline | P. falciparum 80S Ribosome |
| Human Metatastic Cancers |
BL. Lokeshwar, 2011, Pharmacol. Res., 63, 146-150. |
Tetracyclines | Human 80S Ribosome |
About 25% of all patients with GBS develop weakness of the breathing muscles leading to the inability to breathe ade- quately to maintain healthy levels of oxygen in the blood, thus requiring artificial ventilation as in the COVID-related respi- ratory distress. Other complications include venous thrombo- embolism, pneumonia and blood clots in the lungs [41]. Treatment consists of anticoagulants and intravenous immuno- globulins. Seven to ten days (or more) before the onset of GBS, the majority of patients have developed a preceding respiratory or gastrointestinal illness, which may be either mild or completely asymptomatic. Serological studies have shown that Campylobacter jejuni, Epstein Bar virus, and Cytomegalovirus are the most frequent antecedent infections. Kawasaki disease is a disease in which blood vessels throughout the body become inflamed [42]. The most common symptoms include a fever that lasts for more than five days and bilateral conjunc- tival inflammation, followed by mouth symptoms such as red tongue [42]. Other symptoms include sore throat, diarrhea, vomiting and abdominal pain [42]. Initial treatment consists of high doses of aspirin and immunoglobulin. Aspirin, the non-steroidal anti-inflammatory drug (NSAID) is used to reduce inflammation and inhibit platelet aggregation. The specific cause of Kawazaki disease is unknown. The fact that blood clot formation and/or inflammation are common features of these diseases and COVID-19 implies that thromboxanes are likely to be involved in these processes. Thromboxane A2 (TXA2) [43] is produced by activated platelets and presents prothrom- botic properties. It stimulates platelet aggregation by activating the thromboxane receptor. Formation and strengthening of a clot occur when circulating fibrinogen binds to the receptors on adjacent platelets. TXA2 is also a known vasoconstrictor and is especially important during tissue injury and inflammation. TXA2 is generated from prostaglandin H2 by thromboxane-A synthase. Prostaglandin H2 is one of the end-products of the prostaglandins synthesis pathway [43]. Cyclooxygenase 1 (COX1), also known as prostaglandin synthase 1 (PTGS1) or prostaglandinendoperoxide synthase 1, is one of two cyclooxy- genases in humans (COX-1 and COX-2) [43]. It catalyzes the formation of an endoperoxide derivative of arachidonic acid, which represents the first reaction of the prostaglandins synthesis pathway. COX-1 is inhibited by Non-steroidal Anti-Inflammatory Drugs (NSAIDs) such as aspirin. The inhibition of COX-1 by aspirin is irreversible (hence efficient) [43]. Therefore, one can explain why aspirin, with its anticoagulant and anti-inflammatory properties is suitable for the treatment of the Kawazaki and the Guillain-Barré diseases [41, 42]. Since the specific causes of the Kawazaki or the Guillain-Barré diseases are unknown [41, 42], we propose that the patients with COVID-19 or these diseases be treated with individual antibiotics or their combination which are susceptible to abolish the synthesis of all the enzymes of the coagulation and the inflammation processes such as COX-1 and COX-2. The reasoning is that, whatever the infectious agent, the antibiotics selected in the present report are capable of directly inhibiting the ribosomes of the bacteria involved, such as Campylobacter jejuni, or those of the human host cells infected by any infectious agent, so as to prevent the latter from synthesizing the viral proteins and enzymes. Therefore, in any case, we recommend a combination of azithromycin or erythromycin (or equivalent) and tetracycline (or equivalent) for adult patients. In the case of Kawazaki disease, we specifically recommend a combination of josamycin (a macrolide for children) and tetracycline (or equivalent).
CONCLUSION
The study described in the present report has led to the following observations and demonstrations: (i) The discovery (if any) of clinically approved vaccines against SARS-CoV-2 does not make it optional to search for specific and effective therapeutic drugs for COVID-19, as many people are susceptible to be concerned by contraindications. Therefore, in addition to designing and developing vaccines for SARS-CoV-2, repositioning of old drugs such as antibiotics and the development of new drugs are also needed; (ii) It is widely accepted that viral infections cannot be cured by antibiotics whereas bacterial infections can, because the virus has no protein synthesis machinery to produce from its RNA genome, the viral proteins and enzymes essential to its replication and/or to the assembly of viral particles; (iii) As a consequence, in the SARS-CoV-2-infected host cells, the main target for the therapeutically useful drugs is the human protein synthesis machinery built up with the ribosomes, because the virus is forced to rely on the infected host cell to synthesize new proteins that it does not need, but are needed for the virus to spread; (iv) In this regard, it is well established that antibiotics that block the translation process in mammalian cells have potential as therapeutics for diseases such as cancer, inflammation or COVID-19. Therefore, repurposing adequate antibiotics to cure COVID-19 is a highly attractive approach; (v) We propose to reposition the macrolides (azithromycin or erythromycin or others) and tetracyclines for the treatment of COVID-19 patients. Interestingly, these antibiotics had been previously successfully used in medicine in humans and animals. In addition, the binding sites of these antibiotics at the upper part of the protein exit tunnel (for the macrolides) and the ribosomal A-site (for tetracyclines) are universal features of the ribosomes in all kingdoms of life. So, these classes of antibiotics referred to as universal antibiotics are expected to bind to all kinds of ribosomes, the 70S as well as the 80S type, with comparable affinity; (vi) efficacy assessment of these antibiotics in clinical application consisted in comparing their affinity constants of binding to the human ribosomes with their blood concentration. Finally, zinc cations were previously shown to be a strong antiviral agent, while all the macrolides and tetracyclines that we propose for repurposing or repositioning to cure COVID-19 are shown in the present report to form Zn++-antibiotic complex and behave as efficient zinc transporters into the SARS-CoV-2-infected host cells. Therefore, these antibiotics are candidates for specific and effective therapeutic drugs available for COVID-19. In conclusion, we propose to combat the current COVID-19 pandemic with azithromycin or erythromycin (or equivalent) alone or in combination with tetracycline (or equivalent) in the presence of Zn++(SO4--). This treatment should be immediately administered at the onset of COVID-19, upon the first manifestation among the following: fatigue, fever, dry cough, headache, sore throat, muscle pain or shortness of breath. This treatment is susceptible to prevent the synthesis of the cytokines, the glycoproteins (IL-1, IL-6, etc) responsible for the so-called cytokine release storms, the hyperinflammation process leading to the COVID-related Acute Respiratory Distress Syndrome (ARDS). Finally, this treament is susceptible to prevent the blood clot formation and inflammation processes characteristic of the Kawazaki and the Guillain-Barré diseases that are seemingly associated with COVID-19, by the inhibition of the synthesis of cyclooxygenases (COX-1 and COX-2), and hence the formation of thromboxane A2, in order to prevent inflammation and thromboxane A2-mediated contraction in pulmonary veins. In case of Kawazaki disease that concerns the children of 4-5 years, we specifically recommend a combination of josamycin (a macrolide usually administered to children) and tetracycline (or equivalent) in the presence of Zn++(SO4--). As a conclusion for this study, from a clinical viewpoint, the experimental data presented here do not suggest virucidal activity of azithromycin-zinc or tetracycline-zinc complexes. However, they do indicate that when administered simultaneously at the onset of first signs of COVID-19, the most common symptoms being fatigue, fever, dry cough, headache, sore throat, muscle pain or shortness of breath, azithromycin (or tetracycline) and zinc cations are capable of inhibiting ribosomal activity of SARS-CoV-2-infected human cells. This results in blocking protein and enzyme synthesis vital for viral RNA replication and for assembly of viral particles. Although already infected, as shown by the presence of at least one symptom, early treatment allows both reduction (or at worst a non-increase) of viremia as well as stabilizing symptoms. The major advantage of this therapeutic strategy is avoiding prolonged clinical COVID-19 disease with contingent worsening of illness and subsequent need for intensive care. Prolonged COVID-19 illness is the major downfall of the present pandemic, returning to normal being long, difficult, and sometimes impossible. Successful early therapy relies on the rapid detection of disease. During the latent interval between testing and final results, clinical symptoms worsen. The antigenic test allows a rapid diagnosis (taking less than one hour to complete), whereas the PCR test is slower, often requiring several days between patient testing and the final laboratory report.
LIST OF ABBREVIATIONS
| RdRp | = RNA-dependent RNA polymerase |
| PTC | = Peptidyl transferase Center |
| rp | = Ribosomal protein |
| bL12 | = The new name of eubacterial large subunit ribosomal protein L12 with a free NH2 terminus, or its NH2-terminally acetylated form L7 (formerly L7/L12) |
| eL42 | = eukaryal or archaeal large subunit ribosomal protein L42 (formerly L42A or L42AB in yeast or L36a in human, or L44e in archaea) |
| Tet | = Tetracycline |
| IC50 or KIa | = The concentration of antibiotic corresponding to 50% Inhibition of the activity of the human 80S ribosomes |
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
HUMAN AND ANIMAL RIGHTS
No animals/humans were used for studies that are the basis of this research.
CONSENT FOR PUBLICATION
Not applicable.
AVAILABILITY OF DATA AND MATERIALS
Not applicable.
FUNDING
None.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
We are grateful to Prof. Fernand Gbaguidi, Jean Cognet and Jean-Marie Schmitter, as well as to Drs. Urbain C. Kassehin and Giovambattista Scarfone (Cardiology Center Bianco, Italy) for their constant interest in this work, and for fruitful discussions. We gratefully thank Drs. Blanche Aguida, Fulbert Agbo’Saga, Horrhus Hounguè, Soria Baouz-Drahy, Joël Pothier and Gérard Keith for fruitful discussions and for critical reading of the manuscript.

